
INVOKANA 100 mg, comprimé pelliculé, boîte de 30 plaquettes thermoformées de 1
Dernière révision : 05/09/2024
Taux de TVA : 2.1%
Prix de vente : 38,29 €
Taux remboursement SS : 65%
Base remboursement SS : 38,29 €
Laboratoire exploitant : MENARINI FRANCE
Source :

Invokana est indiqué dans le traitement des adultes atteints de diabète de type 2 insuffisamment contrôlé, en complément du régime alimentaire et de l'exercice physique :
- En monothérapie quand la metformine est considérée comme étant inappropriée en raison d'une intolérance ou de contre-indications
- En complément d'autres médicaments pour le traitement du diabète.
Concernant les résultats d'études vis-à-vis des associations de traitements, des effets sur le contrôle glycémique des événements cardiovasculaires et rénaux et des populations étudiées, voir les rubriques Mises en garde spéciales et précautions d'emploi, Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacodynamiques.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Insuffisance rénale
L'efficacité de la canagliflozine pour le contrôle glycémique dépend de la fonction rénale : l'efficacité est moindre chez les patients présentant une insuffisance rénale modérée et probablement absente chez les patients présentant une insuffisance rénale sévère (voir rubrique Posologie et mode d'administration).
Chez les patients avec un DFGe < 60 mL/min/1,73 m² ou une ClCr < 60 mL/min, une incidence plus élevée d'effets indésirables liée à une déplétion volémique (par exemple, sensation vertigineuse posturale, hypotension orthostatique, hypotension) a été rapportée, particulièrement avec la dose de 300 mg. En outre, chez ces patients, davantage d'événements de type hyperkaliémie et des augmentations plus importantes de la créatininémie et de l'urémie (BUN) ont été observés (voir rubrique Effets indésirables).
Par conséquent, la dose de canagliflozine doit être limitée à 100 mg une fois par jour chez les patients avec un DFGe < 60 mL/min/1,73 m² ou une ClCr < 60 mL/min (voir rubrique Posologie et mode d'administration).
Indépendamment du DFGe avant l'instauration du traitement, les patients sous canagliflozine présentent une chute initiale du DFGe qui s'atténue avec le temps (voir rubriques Effets indésirables et Propriétés pharmacodynamiques).
Un suivi de la fonction rénale est recommandé comme suit :
- Avant l'instauration du traitement par la canagliflozine, puis au moins une fois par an par la suite (voir rubriques Posologie et mode d'administration, Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques)
- Avant l'instauration de traitements médicamenteux concomitants susceptibles de diminuer la fonction rénale, puis régulièrement ensuite.
Il existe des données cliniques avec la canagliflozine pour le traitement de la maladie rénale chronique (DFGe≥30 mL/min/1,73 m²) avec ou sans albuminurie. Bien que les deux groupes de patients aient bénéficié du traitement par la canagliflozine, ceux avec albuminurie pourraient en bénéficier davantage.
Utilisation chez les patients présentant un risque d'effets indésirables liés à une déplétion volémique
En raison de son mécanisme d'action (augmentation de l'excrétion urinaire du glucose), la canagliflozine induit une diurèse osmotique, qui peut réduire le volume intravasculaire et diminuer la pression artérielle (voir rubrique Propriétés pharmacodynamiques). Dans les études cliniques contrôlées de la canagliflozine, les augmentations des effets indésirables liés à une déplétion volémique (par exemple, sensation vertigineuse posturale, hypotension orthostatique ou hypotension) ont été plus fréquentes à la dose de 300 mg et se sont produites plus fréquemment au cours des trois premiers mois (voir rubrique Effets indésirables).
Il faut être prudent chez les patients pour lesquels une baisse de la pression artérielle induite par la canagliflozine pourrait présenter un risque, tels que les patients atteints d'une maladie cardiovasculaire, les patients avec un DFGe < 60 mL/min/1,73m2, les patients sous traitement anti- hypertenseur avec antécédent d'hypotension, les patients sous diurétiques, ou les patients âgés (≥ 65 ans) (voir rubriques Posologie et mode d'administration et Effets indésirables).
En raison de la déplétion volémique, des diminutions moyennes généralement faibles du DFGe ont été observées au cours des 6 premières semaines suivant l'instauration du traitement par la canagliflozine. Chez les patients mentionnés ci-dessus, susceptibles de présenter des diminutions plus importantes du volume intravasculaire, on a parfois observé des diminutions plus importantes du DFGe (> 30 %), qui se sont ensuite améliorées et ont peu fréquemment nécessité l'interruption du traitement par la canagliflozine (voir rubrique Effets indésirables).
Il est
conseillé aux patients de signaler les symptômes liés à une déplétion volémique.
La canagliflozine n'est pas recommandée chez les patients
traités par diurétiques de l'anse (voir rubrique Interactions avec d'autres
médicaments et autres formes d'interactions) ou qui ont une déplétion volémique, par exemple à cause d'une maladie aiguë (comme
une maladie gastro-intestinale).
Pour les patients recevant de la canagliflozine, en cas de pathologies intercurrentes pouvant entraîner une déplétion volémique (comme une maladie gastro-intestinale), une surveillance attentive de la volémie (par exemple, examen clinique, mesures de la pression artérielle, bilans biologiques incluant la mesure de la fonction rénale) et du bilan sanguin électrolytique est recommandée. Lorsqu'un patient développe une déplétion volémique sous canagliflozine, l'arrêt temporaire du traitement par la canagliflozine peut être envisagé jusqu'à la correction de cet état. En cas d'arrêt du traitement, il convient de surveiller plus souvent la glycémie.
Acidocétose diabétique
De rares cas d'acidocétose diabétique (ACD), incluant des cas menaçant le pronostic vital ainsi que des cas fatals, ont été rapportés chez des patients traités par des inhibiteurs du SGLT2, dont la canagliflozine. Dans certains cas, la symptomatologie était atypique avec une augmentation seulement modérée de la glycémie, inférieure à 14 mmol/L (250 mg/dL). On ne sait pas si le risque de survenue d'une ACD est plus élevé avec des doses plus fortes de canagliflozine. Le risque d'ACD semble être plus élevé chez les patients présentant une insuffisance rénale modérée à sévère qui ont besoin d'insuline.
Le risque d'acidocétose diabétique doit être envisagé en présence de symptômes non spécifiques tels que nausées, vomissements, anorexie, douleur abdominale, soif excessive, difficultés à respirer, confusion, fatigue inhabituelle ou somnolence. En cas d'apparition de ces symptômes, une acidocétose doit immédiatement être recherchée, quelle que soit la glycémie du patient.
Chez les patients chez lesquels une ACD est suspectée ou a été diagnostiquée, le traitement par Invokana doit immédiatement être arrêté.
Le traitement doit être interrompu chez les patients qui sont hospitalisés pour des pathologies aiguës graves.
Interrompre le traitement par Invokana, si possible, pendant une
période de temps appropriée (jours) avant toute intervention
chirurgicale lourde y compris abdominale et bariatrique, ou tout autre
procédure invasive associée à un jeûne prolongé. La surveillance des
corps cétoniques dans le sang est recommandée. Un traitement
hypoglycémiant alternatif est à envisager, y compris l’insuline.
Le contrôle de la cétonémie (taux de cétone dans le sang) est préféré à la cétonurie (taux de cétone dans l'urine). Le traitement par Invokana peut être repris quand les taux de corps cétoniques sont normaux et l'état du patient stabilisé.
Avant d'instaurer un traitement par Invokana, les antécédents du patient pouvant prédisposer à l'acidocétose doivent être pris en considération.
L'acidocétose diabétique peut être prolongée après l'arrêt de Invokana chez certains patients, c'est-à- dire qu'elle peut durer plus longtemps que prévu compte tenu de la demi-vie plasmatique de la canagliflozine (voir rubrique Propriétés pharmacocinétiques). Une glucosurie prolongée a été observée avec une ACD persistante. Des facteurs indépendants de la canagliflozine pourraient être impliqués dans des périodes prolongées d'ACD. Une carence en insuline peut contribuer à une acidocétose diabétique prolongée et doit être corrigée lorsqu'elle est identifiée.
Les patients susceptibles de présenter un risque accru d'ACD incluent les patients avec une faible réserve fonctionnelle de cellules bêta (par exemple les patients atteints de diabète de type 2 ayant un faible taux de peptide C ou atteints de diabète auto-immun latent de l'adulte (LADA) ou les patients avec un antécédent de pancréatite), les patients avec des affections entraînant une diminution de la prise alimentaire ou une déshydratation sévère, les patients pour lesquels les doses d'insuline sont réduites et les patients ayant des besoins accrus en insuline en raison d'une maladie aiguë, d'une intervention chirurgicale ou d'une consommation excessive d'alcool. Les inhibiteurs du SGLT2 doivent être utilisés avec prudence chez ces patients.
La reprise d'un traitement par inhibiteur du SGLT2 chez les patients avec un antécédent d'ACD au cours d'un traitement par inhibiteur du SGLT2 n'est pas recommandée, à moins qu'un autre facteur déclenchant ait pu être clairement identifié et corrigé.
La sécurité et l'efficacité de la canagliflozine chez les patients atteints de diabète de type 1 n'ont pas été établies et la canagliflozine ne doit pas être utilisée pour le traitement de patients atteints de diabète de type 1. Les données limitées issues des études cliniques suggèrent que l'ACD survient fréquemment lorsque des patients atteints de diabète de type 1 sont traités par des inhibiteurs du SGLT2.
Amputations des membres inférieurs
Dans les études cliniques à long terme utilisant la canagliflozine chez des patients atteints de diabète de type 2 et souffrant d'une maladie cardiovasculaire (MCV) établie ou présentant au moins 2 facteurs de risque de MCV, Invokana était associé a un risque accru d'amputation des membres inférieurs par rapport au placebo (0,63 contre 0,34 événements pour 100 patients-années, respectivement) et cette augmentation survenait principalement pour l'orteil et le médio pied (voir rubrique Effets indésirables). Dans une étude clinique à long terme chez des patients atteints de diabète de type 2 et de maladie rénale chronique, aucune différence du risque d'amputation des membres inférieurs n'a été observée chez les patients traités par canagliflozine 100 mg par rapport au placebo. Dans cette étude, les mesures de précaution indiquées ci-dessous ont été appliquées. Comme le mécanisme sous-jacent n'a pas été mis en évidence, les facteurs de risque d'amputation, hormis les facteurs de risque généraux, sont inconnus.
Avant d'initier un traitement par Invokana, il est nécessaire de prendre en compte les facteurs qui, dans les antécédents du patient, peuvent augmenter le risque d'amputation. À titre de mesures de précaution, il convient de surveiller attentivement les patients présentant un risque d'amputation plus élevé et de conseiller les patients sur l'importance des soins de routine préventifs du pied et du maintien d'une hydratation adéquate. Il convient également d'arrêter le traitement par Invokana chez les patients qui développent des événements susceptibles de précéder une amputation tels qu'un ulcère cutané au niveau des membres inférieurs, une infection, une ostéomyélite ou une gangrène.
Fasciite nécrosante du périnée (gangrène de Fournier)
Des cas de fasciite nécrosante du périnée (aussi appelée «gangrène de Fournier») survenus après mise sur le marché ont été rapportés chez des patients de sexe masculin et féminin prenant des inhibiteurs du SGLT2. Cet événement rare mais grave et mettant potentiellement en jeu le pronostic vital des patients nécessite une intervention chirurgicale et un traitement antibiotique en urgence.
Il convient de recommander aux patients de consulter un médecin s'ils développent des symptômes tels qu'une douleur, une sensibilité, un érythème ou une tuméfaction au niveau de la zone génitale ou périnéale, accompagnés de fièvre ou de malaises. Il convient de garder à l'esprit que la fasciite nécrosante peut être précédée d'une infection urogénitale ou d'un abcès périnéal. En cas de suspicion de gangrène de Fournier, le traitement par Invokana doit être interrompu et un traitement rapide (comprenant des antibiotiques et un débridement chirurgical) doit être instauré.
Hématocrite élevé
Une élévation de l'hématocrite a été observée avec le traitement par canagliflozine (voir rubrique Effets indésirables) ; par conséquent, une surveillance attentive est recommandée chez les patients ayant déjà un hématocrite élevé.
Patients âgés
Les patients âgés peuvent avoir un risque accru de déplétion volémique, sont plus susceptibles d'être traités par des diurétiques et de présenter une insuffisance rénale. Chez les patients âgés de 75 ans et plus, on a observé une incidence plus élevée d'effets indésirables liés à la déplétion volémique (par exemple, sensation vertigineuse posturale, hypotension orthostatique, hypotension). De plus, des diminutions plus importantes du DFGe ont été rapportées chez ces patients (voir rubriques Posologie et mode d'administration et Effets indésirables).
Infections mycosiques génitales
L'apparition de candidoses vulvovaginales chez les femmes et de balanites ou de balanoposthites chez les hommes a été rapportée dans les études cliniques utilisant la canagliflozine, du fait de l'inhibition des co-transporteurs de sodium-glucose de type 2 (SGLT2) entrainant une augmentation de l'excrétion urinaire de glucose (voir rubrique Effets indésirables). Les hommes et les femmes ayant des antécédents d'infections mycosiques génitales ont été plus susceptibles de développer une infection. Les balanites ou les balanoposthites sont principalement apparues chez des hommes non circoncis ce qui, dans certains cas, a entraîné des phimosis et/ou une circoncision. La majorité des infections mycosiques génitales ont été traitées avec des traitements antifongiques topiques, prescrits par un professionnel de santé ou auto-administrés, tout en poursuivant le traitement par Invokana.
Infection des voies urinaires
Des cas d'infection des voies urinaires compliquée, y compris de pyélonéphrite et de sepsis urinaire, survenus après mise sur le marché ont été rapportés chez des patients traités par canagliflozine et ont fréquemment mené à l'interruption du traitement. Une interruption temporaire de la canagliflozine doit être envisagée chez les patients présentant une infection des voies urinaires compliquée.
Insuffisance cardiaque
L'expérience est limitée chez les patients de classe III selon la NYHA (classification de la New York Heart Association) ; par ailleurs, la canagliflozine n'a fait l'objet d'aucune étude clinique chez des patients de classe IV selon la NYHA.
Bilan urinaire
En raison de son mécanisme d'action, les patients prenant de la canagliflozine auront un test de glucose urinaire positif.
Intolérance au lactose
Les comprimés contiennent du lactose. Les patients atteints de troubles héréditaires rares d'intolérance au galactose, de déficit total en lactase, ou de malabsorption du glucose-galactose ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c'est à dire qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
La sécurité de la canagliflozine a été évaluée chez 22 645 patients atteints de diabète de type 2, dont 13 278 patients traités par canagliflozine et 9 367 par un comparateur lors de 15 études cliniques contrôlées de phase 3 et 4 menées en double-aveugle. Au total, 10 134 patients ont été traités dans le cadre de deux études cardiovasculaires dédiées, pendant une durée moyenne d'exposition de149 semaines (223 semaines dans l'essai CANVAS et 94 semaines dans l'essai CANVAS-R) et 8 114 patients ont été traités lors de 12 études cliniques contrôlées de phase 3 et 4 menées en double- aveugle pendant une durée moyenne d'exposition de 49 semaines. Dans une étude dédiée du devenir de la fonction rénale , un total de 4 397 patients atteints de diabète de type 2 et de maladie rénale chronique ont eu une durée moyenne d'exposition de 115 semaines.
L'évaluation principale de la sécurité et de la tolérance a été effectuée dans une analyse poolée (n = 2 313) de quatre études cliniques contrôlées versus placebo de 26 semaines (en monothérapie et en association à metformine, à metformine + sulfamide hypoglycémiant et à metformine + pioglitazone). Les effets indésirables les plus fréquemment rapportés pendant le traitement ont été l'hypoglycémie, lors de l'association à l'insuline ou à un sulfamide hypoglycémiant, les candidoses vulvovaginales, les infections des voies urinaires, ainsi que la polyurie ou la pollakiurie (mictions plus abondantes et plus fréquentes). Les effets indésirables conduisant à l'arrêt du traitement chez ≥ 0,5 % de l'ensemble des patients traités par la canagliflozine dans ces études ont été des candidoses vulvovaginales (0,7 % des femmes traitées), ainsi que des balanites ou des balanoposthites (0,5 % des hommes traités). D'autres analyses de sécurité (incluant des données à long terme) ont été effectuées sur les données correspondant à l'ensemble du programme d'études de la canagliflozine (études contrôlées versus placebo et versus comparateur actif) pour évaluer les effets indésirables rapportés, afin d'identifier les effets indésirables (tableau 2) (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Tableau des effets indésirables
Les effets indésirables présentés dans le tableau 2 sont issus des analyses poolées des études menées versus placebo et comparateur actif décrites ci-dessus. Les effets indésirables rapportés par l'utilisation post-commercialisation de la canagliflozine dans le monde entier sont aussi inclus dans ce tableau. Les effets indésirables mentionnés ci-dessous sont classés par fréquence et par classe de systèmes d'organes. Les différentes catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 2 : Tableau des effets indésirables (MedDRA) à partir des études contrôlées versus placeboe et comparateur actif ainsi que de l'expérience post-commercialisation
| Classe de systèmes d'organes Fréquence | Effet indésirable |
| Infections et infestations | |
| Très fréquent Fréquent Indéterminée | Candidose vulvovaginaleb,j Balanite ou balanoposthiteb,k, Infection des voies urinairesc (pyélonéphrite et sepsis urinaire ont été rapportés après la commercialisation) Fasciite nécrosante du périnée (gangrène de Fournier)d |
| Affections du système immunitaire | |
| Rare | Réaction anaphylactique |
| Troubles du métabolisme et de la nutrition | |
| Très fréquent Peu fréquent Rare | Hypoglycémie en association à l'insuline ou à un sulfamide hypoglycémiantc Déshydratationa Acidocétose diabétiqueb |
| Affections du système nerveux | |
| Peu fréquent | Sensation vertigineuse posturalea, Syncopea |
| Affections vasculaires | |
| Peu fréquent | Hypotensiona, Hypotension orthostatiquea |
| Affections gastro-instestinales | |
| Fréquent | Constipation, Soiff, Nausées |
| Affections de la peau et du tissu sous-cutané | |
| Peu fréquent Rare | Photosensibilité, Éruption cutanéeg, Urticaire Angiœdème |
| Affections musculo-squelettiques et systémiques | |
| Peu fréquent | Fracture osseuseh |
| Affection du rein et des voies urinaires | |
| Fréquent Peu fréquent | Polyurie ou Pollakiuriei Insuffisance rénale (principalement dans le contexte de déplétion volémique) |
| Investigations | |
| Fréquent Peu fréquent | Dyslipidémiel, Hématocrite augmenté,b,m Créatininémie augmentéeb,n, Urémie augmentéeb,o, kaliémie augmentéeb,p, Phosphatémie augmentéeq |
| Actes médicaux et chirurgicaux | |
| Peu fréquent | Amputation des membres inférieurs (principalement l'orteil et du médio pied), en particulier chez les patients à haut risque demaladie cardiaqueb |
b Voir rubrique Mises en garde spéciales et précautions d'emploi et la description de l'EI ci-dessous.
c Voir la description de l'EI ci-dessous.
d Voir rubrique Mises en garde spéciales et précautions d'emploi
e Les profils de données de sécurité des études pivot individuelles (y compris les études menées chez des patients ayant une insuffisance rénale modérée, les patients plus âgés [> 55 ans à < 80 ans], les patients ayant un risque CV et rénal plus élevé) ont généralement corroboré les effets indésirables présentés dans ce tableau.
f Soif inclut les termes soif, sécheresse buccale et polydipsie.
g Éruption cutanée inclut les termes éruption érythémateuse, éruption généralisée, éruption maculeuse, éruption maculopapuleuse, éruption papuleuse, éruption prurigineuse, éruption pustulaire et éruption vésiculaire.
h Lié à une fracture osseuse ; voir la description de l'EI ci-dessous..
i Polyurie ou pollakiurie incluent les termes polyurie, pollakiurie, mictions impérieuses, nycturie et augmentation du volume urinaire.
j Candidose vulvovaginale inclut les termes candidose vulvovaginale, infection mycosique vulvovaginale, vulvovaginite, infection vaginale, vulvite et infection génitale fongique.
k Balanite ou balanoposthite incluent les termes balanite, balanoposthite, balanite à Candida et infection génitale fongique.
l Le pourcentage moyen d'augmentation par rapport à la valeur initiale pour canagliflozine 100 mg et 300 mg versus placebo, était respectivement de 3,4% et 5,2% versus 0,9% pour le cholestérol total ; 9,4% et 10,3% versus 4,0% pour le HDL-cholestérol ; 5,7% et 9,3% versus 1,3% pour le LDL-cholestérol ; 2,2% et 4,4% versus 0,7% pour le cholestérol non-HDL ; 2,4% et 0.0% versus 7,6% pour les triglycérides.
m La variation moyenne de l'hématocrite par rapport à la valeur initiale était respectivement de 2,4% et 2,5% pour canagliflozine 100 mg et 300 mg, comparée à 0,0% pour le placebo.
n Le pourcentage moyen de variation de la créatinine par rapport à la valeur initiale était respectivement de 2,8% et 4,0% pour canagliflozine 100 mg et 300 mg comparé à 1.5% pour le placebo.
o Le pourcentage moyen de variation de l'urémie par rapport à la valeur initiale était respectivement de 17,1% et 18,0% pour canagliflozine 100 mg et 300 mg, comparé à 2,7% pour le placebo.
p Le pourcentage moyen de variation de la kaliémie par rapport à la valeur initiale était respectivement de 0,5% et 1,0% pour canagliflozine 100 mg et 300 mg, comparé à 0,6% pour le placebo.
q Le pourcentage moyen de variation de la phosphatémie par rapport à la valeur initiale était respectivement de 3,6% et 5,1% pour canagliflozine 100 mg et 300 mg, comparé à 1,5% pour le placebo.
Description de certains effets indésirables
Acidocétose diabétique
Dans une étude du devenir de la fonction rénale à long terme chez des patients atteints de diabète de type 2 et de maladie rénale chronique, les taux d'incidence des événements avérés d'acidocétose diabétique (ACD) étaient de 0,21 (0,5 %, 12/2 200) et de 0,03 (0,1 %, 2/2 197) pour 100 patients- années de suivi avec la canagliflozine 100 mg et le placebo, respectivement ; parmi les 14 patients atteints d'ACD, 8 (7 sous canagliflozine 100 mg et 1 sous placebo) avaient un DFGe avant traitement, compris entre 30 et < 45 mL/min/1,73 m2 (voir rubrique Mises en garde spéciales et précautions d'emploi).
Amputation des membres inférieurs
Chez les patients souffrant de diabète de type 2 et présentant une maladie cardiovasculaire établie ou au moins 2 facteurs de risque de maladie cardiovasculaire, la canagliflozine a été associée à une augmentation du risque d'amputation des membres inférieurs, tel qu'observé dans le programme intégré CANVAS, comprenant CANVAS et CANVAS-R, deux essais de grande ampleur, de longue durée, randomisés, contrôlés contre placebo visant à évaluer 10 134 patients. Le déséquilibre s'est produit dés les 26 premières semaines de traitement. Les patients des essais CANVAS et CANVAS-R ont été suivis pendant une moyenne de respectivement 5,7 et 2,1 ans. Indépendamment du fait que le traitement soit la canagliflozine ou le placebo, le risque d'amputation était supérieur chez les patients ayant un antécédent d'amputation, de maladie vasculaire périphérique ou de neuropathie. Le risque d'amputation des membres inférieurs n'était pas dose-dépendant. Les résultats relatifs à l'amputation du programme intégré CANVAS sont présentés par le tableau 3.
Il n'y avait pas de différence de risque d'amputation des membres inférieurs associé à l'utilisation de canagliflozine 100 mg par rapport au placebo (1,2 contre 1,1 événements pour 100 patients-années, respectivement [RR : 1,11 ; IC 95 % 0,79, 1,56]) dans l'étude CREDENCE, une étude du devenir de lafonction rénale à long terme de 4 397 patients atteints de diabète de type 2 et de maladie rénale chronique (voir rubrique Mises en garde spéciales et précautions d'emploi). Dans d'autres études sur le diabète de type 2 portant sur la canagliflozine, incluant une population diabétique générale de 8 114 patients, aucune différence n'a été observée par rapport au contrôle vis-à-vis du risque d'amputation des membres inférieurs.
Tableau 3 : Analyse intégrée des amputations dans les essais CANVAS ET CANVAS-R
| Placebo N = 4 344 | canagliflozine N = 5 790 | |
| Nombre total de sujets présentant desévénements, n (%) | 47 (1,1) | 140 (2,4) |
| Taux d'incidence (pour 100 patients-années) | 0,34 | 0,63 |
| RR (IC 95 %) vs. placebo | 1,97 (1,41, 2,75) | |
| Amputation mineure, n (%)* | 34/47 (72,3) | 99/140 (70,7) |
| Amputation majeure, n (%)† | 13/47 (27,7) | 41/140 (29,3) |
* Orteil et médio pied
† Cheville, sous le genou et au-dessus du genou
Dans le programme CANVAS, chez les sujets ayant subi une amputation, l'orteil et le médio pied représentaient les sites les plus fréquemment touchés (71%) dans les deux groupes de traitement (tableau 3). Des amputations multiples (certaines impliquant les deux membres inférieurs) ont été observées peu fréquemment et dans des proportions similaires au sein des deux groupes de traitement.
Les infections des membres inférieurs, ulcères du pied diabétique, artériopathies périphériques et gangrènes étaient les événements médicaux les plus fréquemment associés à la nécessité d'une amputation au sein des deux groupes de traitement (voir rubrique Mises en garde spéciales et précautions d'emploi).
Effets indésirables liés à la déplétion volémique
Dans les analyses poolées des quatre études contrôlées versus placebo de 26 semaines, l'incidence de tous les effets indésirables liés à la déplétion volémique (par exemple, sensation vertigineuse posturale, hypotension orthostatique, hypotension, déshydratation et syncope) a été de 1,2 % pour canagliflozine 100 mg, 1,3 % pour canagliflozine 300 mg et 1,1 % pour le placebo. Dans les deux études contrôlées versus traitement actif, l'incidence avec la canagliflozine a été similaire à celle observée avec les comparateurs actifs.
Dans l'une des études cardiovasculaires de longue durée dédiées (CANVAS), dans laquelle les patients étaient généralement plus âgés, avec un taux plus élevé de complications diabétiques, les taux d'incidence des effets indésirables liés à la déplétion volémique ont été de 2,3 événements pour100 patients-années avec canagliflozine 100 mg une fois par jour, 2,9 avec canagliflozine 300 mg et 1,9 avec placebo.
Pour évaluer les facteurs de risque relatifs à ces effets indésirables, une analyse poolée à plus grande échelle (N = 12 441) a été menée chez des patients provenant de 13 études de phase 3 et de phase 4 contrôlées incluant les deux doses de canagliflozine. Dans cette analyse poolée, les patients traités par diurétiques de l'anse, les patients avec un DFGe initial compris entre 30 ml/min/1,73 m2 et < 60 ml/min/1,73 m2 et les patients âgés de 75 ans et plus avaient généralement des incidences supérieures de ces effets indésirables. Pour les patients sous diurétiques de l'anse, les taux d'incidence ont été de 5,0 événements pour 100 patients-années avec canagliflozine 100 mg et 5,7 avec canagliflozine 300 mg, contre 4,1 événements pour 100 patients-années d'exposition dans le groupe contrôle. Pour les patients avec un DFGe initial compris entre 30 ml/min/1,73 m2 et < 60 ml/min/1,73 m2, les taux d'incidence ont été de 5,2 événements pour 100 patients-années d'exposition avec canagliflozine 100 mg et 5,4 avec canagliflozine 300 mg, contre 3,1 événements pour 100 patients-années d'exposition dans le groupe témoin. Chez les patients âgés de 75 ans et plus, les taux d'incidence ont été de 5,3 avec canagliflozine 100 mg et 6,1 avec canagliflozine 300 mg, contre 2,4 événements pour 100 patients-années d'exposition dans le groupe contrôle (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Dans une étude du devenir de la fonction rénale à long terme chez des patients atteints de diabète de type 2 et de maladie rénale chronique, les taux d'incidence des événements liés à la déplétion volémique étaient de 2,84 et 2,35 événements pour 100 patients-années pour canagliflozine 100 mg et le placebo, respectivement. Il a été observé que le taux d'incidence augmentait lorsque le DFGe diminuait. Chez les sujets présentant un DFGe compris entre 30 et <45 mL/min/1,73 m2, le taux d'incidence de la déplétion volémique était plus élevé dans le groupe canagliflozine (4,91 événements pour 100 patients-années) que dans le groupe placebo (2,60 événements pour 100 patients-années) ; toutefois, dans les sous-groupes ayant un DFGe compris entre ≥45 et <60 et compris entre 60et <90 mL/min/1,73 m2, le taux d'incidence entre les groupes était similaire.
Dans l'étude cardiovasculaire dédiée et l'analyse poolée à plus grande échelle, ainsi que dans l'étude dédiée du devenir de la fonction rénale, les arrêts de traitements dus à des effets indésirables liés à la déplétion volémique et à des effets indésirables graves liés à la déplétion volémique n'ont pas augmenté avec la canagliflozine.
Hypoglycémie en cas d'association à l'insuline ou à un sécrétagogue de l'insuline
La fréquence des hypoglycémies a été faible (environ 4 %) dans les différents groupes de traitement, y compris le groupe sous placebo, lorsque la canagliflozine a été utilisée en monothérapie ou en association à la metformine. Lorsque la canagliflozine a été ajoutée à une insulinothérapie, on a observé une hypoglycémie chez respectivement 49,3 %, 48,2 % et 36,8 % des patients traités par canagliflozine 100 mg, canagliflozine 300 mg et placebo et une hypoglycémie sévère s'est produite chez respectivement 1,8 %, 2,7 % et 2,5 % des patients traités par canagliflozine 100 mg, canagliflozine 300 mg et placebo. Lorsque la canagliflozine a été ajoutée à un traitement par sulfamide hypoglycémiant, une hypoglycémie a été observée chez respectivement 4,1 %, 12,5 % et 5,8 % des patients traités par canagliflozine 100 mg, canagliflozine 300 mg et placebo (voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions).
Infections mycosiques génitales
Une
candidose vulvovaginale (incluant une vulvovaginite et une infection
mycosique vulvovaginale) a été observée chez respectivement 10,4 % et
11,4 % des femmes traitées par canagliflozine 100 mg et canagliflozine
300 mg, contre 3,2 % chez les patientes sous placebo. La plupart des
candidoses vulvovaginales sont apparues au cours des quatre premiers
mois de traitement par canagliflozine.
2,3 % des femmes sous canagliflozine ont présenté plus d'une infection.
Dans l'ensemble, 0,7 % des patientes ont arrêté le traitement par
canagliflozine en raison d'une candidose vulvovaginale. La durée des
symptômes était comparable entre le traitement par canagliflozine et le
placebo (voir rubrique Mises en garde spéciales et précautions d'emploi).
Dans le programme CANVAS, les patients souffrant de diabète de type 2
et présentant une maladie cardiovasculaire établie ou au moins 2
facteurs de risques de maladie cardiovasculaire ont subi une durée
d'infection plus longue (voir rubrique Mises en garde spéciales et précautions d'emploi).
Dans le programme CANVAS, la durée médiane de l'infection était plus
longue dans le groupe canagliflozine que dans le groupe placebo.
Une balanite candidosique ou une balanoposthite a affecté les hommes selon un taux de, respectivement, 2,98 et 0,79 événements pour 100 patients-années sous canagliflozine et placebo. Chez les hommes sous canagliflozine, 2,4 % ont présenté plus d'une infection. L'arrêt de la canagliflozine chez les hommes en raison d'une balanite candidosique ou d'une balanoposthite s'est produit selon un taux de 0,37 événements pour 100 patients-années. Des phimosis ont été rapportés selon un taux de, respectivement, 0,39 et 0,07 événements pour 100 patients-années sous canagliflozine et placebo. Une circoncision a été réalisée respectivement à des taux de 0,31 et 0,09 événements pour 100 patients-années sous canagliflozine et placebo (voir rubrique Mises en garde spéciales et précautions d'emploi).
Infections des voies urinaires
Dans les études cliniques, la survenue d'infections des voies urinaires a été plus fréquente sous canagliflozine 100 mg et 300 mg (respectivement 5,9 %, et 4,3 %), comparativement à la fréquence observée sous placebo (4,0 %). La plupart des infections ont été légères à modérées, sans augmentation de l'apparition des effets indésirables graves. Dans ces études, les sujets ont répondu à des traitements standards tout en continuant le traitement par canagliflozine.
Néanmoins, des cas d'infection des voies urinaires compliquée, y compris de pyélonéphrite et de sepsis urinaire, survenus après mise sur le marché ont été rapportés chez des patients traités par canagliflozine et ont fréquemment mené à l'interruption du traitement.
Fracture osseuse
Dans une étude cardiovasculaire (CANVAS) de 4 327 sujets traités pour une maladie cardiovasculaire établie ou présentant au moins deux facteurs de risque de maladie cardiovasculaire, les taux d'incidence de l'ensemble des fractures osseuses avérées étaient respectivement de 1,6, 1,8 et 1,1 événements pour 100 patients-années de suivi, respectivement sous 100 mg de canagliflozine, 300 mg de canagliflozine et placebo, avec un déséquilibre de ce taux survenant initialement dans les26 premières semaines de traitement.
Dans deux autres études à long terme et dans les études effectuées dans la population diabétique générale, aucune différence concernant le risque de fracture n'a été observée avec la canagliflozine par rapport au groupe contrôle. Dans une deuxième étude cardiovasculaire (CANVAS-R) de 5 807 sujets traités pour une maladie cardiovasculaire établie ou présentant au moins deux facteurs de risque de maladie cardiovasculaire, les taux d'incidence de l'ensemble des fractures osseuses avérées étaient respectivement de 1,1 et 1,3 événements pour 100 patients-années de suivi, sous canagliflozine et placebo.
Dans une étude du devenir de la fonction rénale à long terme de 4 397 sujets traités atteints de diabète de type 2 et de maladie rénale chronique, les taux d'incidence de l'ensemble des fractures osseuses avérées étaient de 1,2 événements pour 100 patients-années de suivi avec la canagliflozine 100 mg et avec le placebo. Dans les autres études avec la canagliflozine dans le diabète de type 2, qui ont inclus une population diabétique générale de 7 729 patients et où les fractures osseuses ont été évaluées, les taux d'incidence de l'ensemble des fractures osseuses avérées étaient respectivement de 1,2 et 1,1 événements pour 100 patients-années de suivi, sous canagliflozine et placebo. Après 104 semaines de traitement, la canagliflozine n'a pas affecté la densité minérale osseuse.
Populations particulières
Patients âgés
Dans une analyse poolée de 13 études contrôlées versus placebo et contrôlées versus comparateur actif, le profil de sécurité de canagliflozine chez les patients âgés était généralement cohérent avec celui des patients plus jeunes. Les patients âgés de 75 ans et plus avaient une incidence plus élevée d'effets indésirables liés à la déplétion volémique (comme les sensations vertigineuses posturales, l'hypotension orthostatique, l'hypotension), avec des taux d'incidence respectivement de 5,3, 6,1 et 2,4 événements pour 100 patients-années d'exposition pour canagliflozine 100 mg,canagliflozine 300 mg et le groupe contrôle. Des diminutions du DFGe (-3,4 et -4,7 ml/min/1,73 m2) ont été respectivement rapportées dans les groupes canagliflozine 100 mg et 300 mg, comparativement au groupe contrôle (-4.2 ml/min/1,73 m2). Les DFGe moyens de référence étaient respectivement de 62,5, 64,7 et 63,5 ml/min/1,73 m2 pour la canagliflozine 100 mg, la canagliflozine 300 mg et le groupe contrôle (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Insuffisance rénale chez les patients atteints de diabète de type 2 insuffisamment contrôlé
Les patients avec un DFGe < 60 ml/min/1,73 m2 avaient une incidence supérieure d'effets indésirables associés à la déplétion volémique (par exemple, sensation vertigineuse posturale, hypotension orthostatique, hypotension) avec des taux d'incidence respectivement de 5,3, 5,1 et 3,1 événements pour 100 patients-années d'exposition pour canagliflozine 100 mg, canagliflozine 300 mg et placebo (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Le taux d'incidence globale de potassium sérique élevé était supérieur chez les patients souffrant d'insuffisance rénale modérée, avec des taux d'incidence, respectivement, de 4,9, 6,1 et 5,4 événements pour 100 patients-années d'exposition pour canagliflozine 100 mg, canagliflozine 300 mg et placebo. En général, ces augmentations de la kaliémie ont été transitoires et n'ont pas nécessité de traitement spécifique.
Chez les patients souffrant d'insuffisance rénale modérée, une augmentation de la créatinine sérique de 9,2 µmol/l et de l'azote uréique du sang d'environ 1,0 mmol/l aux deux doses de canagliflozine a été observée.
Les taux d'incidence de plus fortes diminutions du DFGe (> 30 %), à tout moment du traitement, étaient respectivement de 7,3, 8,1 et 6,5 événements pour 100 patients-années d'exposition pour canagliflozine 100 mg, canagliflozine 300 mg et placebo. À la dernière valeur post-référence, les taux d'incidence de ces diminutions étaient de 3,3 pour les patients traités par canagliflozine 100 mg, 2,7 pour canagliflozine 300 mg et 3,7 événements pour 100 patients-années d'exposition pour le placebo (voir rubrique Mises en garde spéciales et précautions d'emploi).
Quel que soit leur DFGe de référence, chez les patients traités par canagliflozine, le DFGe moyen a commencé par chuter. Par la suite, le DFGe s'est maintenu ou a progressivement augmenté durant la suite du traitement. Le DFGe moyen est revenu à la valeur de référence après l'arrêt du traitement, ce qui suggère que des modifications hémodynamiques peuvent jouer un rôle dans ces modifications de la fonction rénale.
Insuffisance rénale chez les patients atteints de maladie rénale chronique dans le diabète de type 2
Dans une étude du devenir de la fonction rénale à long terme chez des patients atteints de diabète de type 2 et de maladie rénale chronique, l'incidence des événements liés à la fonction rénale était élevée dans les deux groupes, mais moins élevée dans le groupe canagliflozine (5,71 événements pour 100 patients-années) que dans le groupe placebo (7,91 événements pour 100 patients-années). Les événements graves et sévères liés à la fonction rénale étaient également moins fréquents dans le groupe canagliflozine que dans le groupe placebo. Les taux d'incidence des événements liés à la fonction rénale étaient inférieurs avec la canagliflozine par rapport au placebo dans les trois strates de DFGe ; le taux d'incidence le plus élevé des événements liés à la fonction rénale a été observé dans la strate où le DFGe est compris entre 30 et < 45 mL/min/1,73 m2 (9,47 contre 12,80 événements pour 100 patients-années respectivement pour la canagliflozine et le placebo).
Dans l'étude du devenir de la fonction rénale à long terme, aucune différence du potassium sérique, aucune augmentation des événements indésirables d'hyperkaliémie, et aucune augmentation absolue (> 6,5 mEq/L) ou relative (> limite supérieure de la normale et > 15 % d'augmentation par rapport aux valeurs initiales) du potassium sérique n'ont été observées avec canagliflozine 100 mg par rapport au placebo.
En
général, aucun déséquilibre n'a été observé entre les groupes de
traitement en ce qui concerne les anomalies du phosphate, dans
l'ensemble ou dans chaque catégorie de DFGe (45 à < 60 ou30 à < 45 mL/min/1,73 m2 [ClCr 45 à < 60 ou 30 à < 45 mL/min]).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via https://signalement.social-sante.gouv.fr.
NE PAS INITIER de traitement par canagliflozine si DFGe (mL/min/1,73 m2) ou ClCr (mL/min) < 30.
AVANT l'instauration du traitement :
- Corriger une déplétion volémique.
- Surveiller la fonction rénale.
- Prendre en considération les antécédents du patient pouvant prédisposer à l'acidocétose.
- Prendre en compte les facteurs qui, dans les antécédents du patient, peuvent augmenter le risque d'amputation.
SURVEILLANCE :
- Fonction rénale, au moins une fois par an.
- Des patients présentant un risque d'amputation plus élevé. Les
conseiller sur l'importance des soins de routine préventifs du pied et
du maintien d'une hydratation adéquate.
INTERFERENCE avec les tests de laboratoire :
En raison de son mécanisme d'action, les patients prenant de la canagliflozine auront un test de glucose urinaire positif.
ARRETER LE TRAITEMENT ET CONSULTER IMMEDIATEMENT UN MEDECIN en cas de :
- Gonflement de la face, des lèvres, de la bouche, de la langue, ou de
la gorge qui peut engendrer une difficulté à respirer ou à avaler.
- Perte de poids rapide, nausées ou vomissements, douleur à l'estomac,
soif excessive, respiration rapide et profonde, confusion, somnolence
ou fatigue inhabituelle, odeur sucrée au niveau du souffle, goût sucré
ou métallique dans la bouche ou modification de l'odeur des urines ou
de la transpiration.
- Perte d'une quantité trop importante de liquides. Les signes
possibles de déshydratation sont une sensation d'étourdissement ou
vertigineuse, une perte de connaissance (évanouissement) ou des
sensations vertigineuses ou une perte de connaissance au moment du
passage en position debout, une bouche très sèche ou collante, une
sensation de soif intense, une sensation de très grande faiblesse ou de
fatigue, une difficulté ou impossibilité d'uriner, un rythme cardiaque
rapide.
INFORMER DES QUE POSSIBLE LE MEDECIN en cas de :
- Signes d'hypoglycémie : vision floue, picotements des lèvres,
tremblements, transpiration, pâleur, changement d'humeur ou sentiment
d'anxiété ou de confusion.
- Fièvre et/ou frissons, sensation de brûlure pendant la miction
(évacuation des urines), douleur dans le dos ou sur les côtés. Sang
dans les urines.
- Lésion, décoloration, ou bien sensibilité ou douleur au niveau des pieds.
- Douleur, sensibilité, rougeur ou tuméfaction au niveau des parties
génitales ou de la zone qui s'étend des parties génitales à l'anus,
accompagnés de fièvre ou d'une sensation générale de malaise.
- Signes d'infection génitale fongique comme une irritation, une démangeaison, un écoulement ou une odeur inhabituels.
VERIFIER régulièrement l'état de ses pieds, en prendre soin et maintenir une hydratation adéquate.
EVITER la prise de préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
PRUDENCE en cas de conduite de véhicules et d'utilisation de machines (sensations vertigineuses, étourdissements).
CONTINUER à suivre les conseils des professionnels de santé en termes de régime alimentaire et d'exercice physique.
DEMANDER au médecin
s'il faut arrêter de prendre le traitement et quand le reprendre en cas d'intervention
chirurgicale lourde ou de procédure nécessitant un jeûne prolongé.
Grossesse
Il n'existe pas de donnée sur l'utilisation de la canagliflozine chez la femme enceinte. Les études chez l'animal ont montré une toxicité de reproduction (voir rubrique Données de sécurité préclinique).
La canagliflozine ne doit pas être utilisée pendant la grossesse. Quand une grossesse est détectée le traitement par la canagliflozine doit être arrêté.
Allaitement
On ne sait pas si la canagliflozine et/ou ses métabolites sont excrétés dans le lait maternel. Les données pharmacodynamiques/toxicologiques disponibles chez l'animal ont mis en évidence l'excrétion de la canagliflozine/métabolites dans le lait, ainsi que des effets pharmacologiques chez la progéniture allaitée des rates et chez les rats juvéniles exposés à la canagliflozine (voir rubrique Données de sécurité préclinique). Un risque pour les nouveaux-nés/nourrissons ne peut être exclu. La canagliflozine ne doit pas être utilisée pendant l'allaitement.
Fertilité
L'effet de la canagliflozine sur la fertilité n'a pas été étudié chez l'Homme. Aucun effet sur la fertilité n'a été observé dans les études chez l'animal (voir rubrique Données de sécurité préclinique).
Interactions pharmacodynamiques
Diurétiques
La canagliflozine peut majorer l'effet des diurétiques et peut augmenter le risque de déshydratation et d'hypotension (voir rubrique Mises en garde spéciales et précautions d'emploi).
Insuline et sécrétagogues de l'insuline
L'insuline et les sécrétagogues de l'insuline, comme les sulfamides hypoglycémiants, peuvent entraîner une hypoglycémie. Par conséquent, une dose plus faible d'insuline ou de sécrétagogue de l'insuline peut être nécessaire pour réduire le risque d'hypoglycémie lorsqu'ils sont utilisés en association avec la canagliflozine (voir rubriques Posologie et mode d'administration et Effets indésirables).
Interactions pharmacocinétiques
Effet des autres médicaments sur la canagliflozine
Le métabolisme de la canagliflozine s'effectue principalement via la glucuronidation médiée par l'UDP glucuronosyl transferase 1A9 (UGT1A9) et 2B4 (UGT2B4). La canagliflozine est transportée par la glycoprotéine P (P-gp) et la BCRP (Breast Cancer Resistance Protein).
Les inducteurs enzymatiques (comme le millepertuis [Hypericum perforatum], la rifampicine, les barbituriques, la phénytoïne, la carbamazépine, le ritonavir, l'éfavirenz) diminuent l'exposition à la canagliflozine. Après la co-administration de canagliflozine et de rifampicine (un inducteur de différents transporteurs actifs et d'enzymes métabolisant les médicaments), des diminutions de respectivement 51 % et 28 % de l'exposition systémique à la canagliflozine (ASC) et de la concentration maximale (Cmax) ont été observées. Ces diminutions d'exposition à la canagliflozine peuvent diminuer son efficacité.
En cas d'administration concomitante d'un médicament inducteur de ces protéines de transport et de ces enzymes UGT avec la canagliflozine, il convient d'instaurer une surveillance du contrôle glycémique pour évaluer la réponse à la canagliflozine. En cas d'administration concomittante d'un médicament inducteur de ces enzymes UGT avec la canagliflozine, il peut être envisagé d'augmenter la dose à 300 mg une fois par jour chez les patients qui tolèrent 100 mg de canagliflozine une fois par jour, avec un DFGe ≥ 60 mL/min/1,73 m2 ou une ClCr ≥ 60 mL/min, et qui nécessitent un contrôle glycémique renforcé. Chez les patient avec un DFGe compris entre 45 mL/min/1,73 m2 et < 60 mL/min/1,73 m2 ou une ClCr comprise entre 45 mL/min et < 60 mL/min traités de manière concomittante par canagliflozine 100 mg et par un inducteur d'enzyme UGT, nécessitant un contrôle glycémique renforcé, d'autres thérapies hypoglycémiantes doivent être envisagées (voir rubrique Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
La cholestyramine peut potentiellement réduire l'exposition à la canagliflozine. La canagliflozine doit être prise 1 heure avant ou 4-6 heures après l'administration d'un chélateur de l'acide biliaire afin de minimiser les interférences possibles avec son absorption.
Des études d'interaction suggèrent que la pharmacocinétique de la canagliflozine n'est pas altérée par la metformine, l'hydrochlorothiazide, les contraceptifs oraux (éthinylestradiol et lévonorgestrel), la ciclosporine et/ou le probénécide.
Effets de la canagliflozine sur les autres médicaments
Digoxine
L'association de canagliflozine à 300 mg une fois par jour pendant 7 jours à une dose unique de 0,5 mg de digoxine, suivie de 0,25 mg par jour pendant 6 jours, a entrainé une augmentation de 20 % de l'ASC et une augmentation de 36 % de la Cmax de la digoxine, en raison d'une inhibition de la P-gp. Il a été observé que la canagliflozine inhibe la P-gp in vitro. Les patients prenant de la digoxine ou d'autres glucosides cardiotoniques (par exemple, digitoxine) doivent être surveillés de manière appropriée.
Lithium
L'utilisation concomitante d'un inhibiteur du SGLT2 avec du lithium peut diminuer les concentrations sériques de lithium. Surveiller plus fréquemment la concentration sérique de lithium pendant le traitement par la canagliflozine, en particulier lors de l'initiation et des changements de posologie.
Dabigatran
L'administration concomittante de canagliflozine (inhibiteur faible de la P-gp) sur le dabigatran étexilate (un substrat de la P-gp) n'a pas été étudiée. Comme les concentrations de dabigatran peuvent être augmentées en présence de canagliflozine, une surveillance (recherche de signes d'hémorragie ou d'anémie) doit être effectuée en cas d'association du dabigatran avec la canagliflozine.
Simvastatine
L'association de canagliflozine 300 mg une fois par jour pendant 6 jours à une dose unique de simvastatine (substrat de CYP3A4) 40 mg a entrainé une augmentation de 12 % de l'ASC et une augmentation de 9 % de la Cmax de la simvastatine et une augmentation de 18 % de l'ASC et une augmentation de 26 % de la Cmax de la simvastatine acide. L'augmentation de l'exposition à la simvastatine et à la simvastatine acide n'est pas considérée comme cliniquement significative.
L'inhibition de la BCRP par la canagliflozine au-niveau de l'intestin ne peut être exclue et une augmentation de l'exposition aux médicaments transportés par la BCRP (par exemple les statines telles que la rosuvastatine et certains agents anti-cancéreux) peut survenir.
Dans des études d'interaction, la canagliflozine à l'état d'équilibre n'a pas eu d'effet cliniquement pertinent sur la pharmacocinétique de la metformine, des contraceptifs oraux (éthinylestradiol, lévonorgestrel), du glibenclamide, du paracétamol, de l'hydrochlorothiazide, ou de la warfarine.
Interférence médicament / test de laboratoire
Test 1,5-AG
L'augmentation de l'excrétion urinaire du glucose (UGE) avec Invokana peut faussement baisser les taux de 1,5-anhydroglucitol (1,5-AG) et rend la mesure du 1,5-AG non fiable pour l'évaluation du contrôle glycémique. Par conséquent, le test 1,5-AG ne doit pas être utilisé pour l'évaluation du contrôle glycémique chez les patients sous canagliflozine. Pour plus de détails, il est conseillé de contacter le fabricant du test 1,5-AG.
Posologie
La
dose initiale de canagliflozine recommandée est de 100 mg une fois par
jour. Chez les patients qui tolèrent la dose de 100 mg de
canagliflozine une fois par jour, et dont le débit de filtration
glomérulaire estimé (DFGe) est ≥ 60 mL/min/1,73 m² ou la ClCr ≥ 60
mL/min et qui nécessitent un contrôle glycémique plus étroit, la dose
peut être augmentée à 300 mg par jour (voir rubrique Mises en garde spéciales et précautions d'emploi).
Pour les recommandations concernant l'adaptation posologique en fonction du DFGe, consulter le tableau 1.
Des précautions doivent être prises lors de l'augmentation de la dose chez les patients âgés de 75 ans et plus, les patients atteints d'une maladie cardiovasculaire, ou les autres patients pour lesquels la diurèse initiale induite par la canagliflozine présente un risque (voir rubrique Mises en garde spéciales et précautions d'emploi). Chez les patients présentant des signes de déplétion volémique, il est recommandé de corriger cet état avant l'instauration du traitement par la canagliflozine (voir rubrique Mises en garde spéciales et précautions d'emploi).
Lorsque la canagliflozine est utilisée en association à l'insuline ou à un sécrétagogue de l'insuline (par exemple les sulfamides hypoglycémiants), une dose plus faible d'insuline ou de sécrétagogue de l'insuline peut être envisagée pour réduire le risque d'hypoglycémie (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Effets indésirables).
Populations particulières
Patients âgés
La fonction rénale et le risque de déplétion volémique doivent être pris en compte (voir rubrique Mises en garde spéciales et précautions d'emploi).
Insuffisance rénale
Pour le traitement de la maladie rénale chronique chez les patients diabétiques de type 2, en complément du traitement standard (par exemple des IEC ou des ARA II), une dose de 100 mg de canagliflozine une fois par jour doit être utilisée (voir tableau 1). Du fait que l'efficacité hypoglycémiante de la canagliflozine est réduite chez les patients présentant une insuffisance rénale modérée et probablement absente chez les patients présentant une insuffisance rénale sévère, si un contrôle glycémique renforcé est nécessaire, l'ajout d'agents anti-hyperglycémiques doit être envisagé. Pour les recommandations concernant l'adaptation posologique en fonction du DFGe, consulter le tableau 1.
Tableau 1 : Recommandations concernant l'adaptation posologiquea
| DFGe (mL/min/1,73 m2) ou ClCr (mL/min) | Dose quotidienne totale de canagliflozine |
| ≥ 60 | Initier avec 100 mg. Chez les patients tolérant 100 mg et nécessitant un contrôle glycémique renforcé, la dose peut être augmentée jusqu'à 300 mg. |
| 30 à < 60b | Utiliser 100 mg. |
| < 30b,c | Continuer avec 100 mg pour les patients prenant déjà Invokanad. Invokana ne doit pas être initié. |
b Si un contrôle glycémique renforcé est nécessaire, l'ajout d'agents anti-hyperglycémiques doit être envisagé.
c Avec un rapport albumine/créatine urinaire ˃ 300 mg/g
d Continuer l'administration jusqu'à la dialyse ou une transplantation rénale
Insuffisance hépatique
Chez les patients atteints d'insuffisance hépatique légère ou modérée, aucune adaptation posologique
n'est nécessaire.
La canagliflozine n'a pas été étudiée chez les patients présentant une insuffisance hépatique sévère et son utilisation n'est pas recommandée chez ces patients (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité de la canagliflozine chez les enfants âgés de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie orale.
Invokana doit être pris par voie orale une fois par jour, de préférence avant le premier repas de la journée. Les comprimés doivent être avalés entiers.
Si une dose est oubliée, elle doit être prise dès que le patient s'en souvient ; cependant, aucune dose double ne doit être prise le même jour.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
L'administration de doses uniques atteignant 1 600 mg de canagliflozine chez des sujets sains et l'administration de 300 mg de canagliflozine deux fois par jour pendant 12 semaines chez des patients atteints de diabète de type 2 ont généralement été bien tolérées.
Traitement
En cas de surdosage, il est raisonnable de recourir aux mesures habituelles, par exemple, éliminer le produit non absorbé du tube digestif, effectuer une surveillance clinique, voire instaurer un traitement symptômatique le cas échéant. La canagliflozine a été très faiblement éliminée par une séance d'hémodialyse de 4 heures. La canagliflozine ne semble pas dialysable par dialyse péritonéale.
Classe pharmacothérapeutique : médicaments utilisés dans le traitement du diabète, médicaments hypoglycémiants, à l'exclusion des insulines. Code ATC : A10BK02.
Mécanisme d'action
Le co-transporteur sodium-glucose de type 2 (SGLT2), exprimé dans les tubules rénaux proximaux, est responsable de la majorité de la réabsorption du glucose filtré depuis la lumière tubulaire. Chez les patients diabétiques, la réabsorption rénale du glucose est augmentée, ce qui peut contribuer à l'élévation persistante de la glycémie. La canagliflozine est un inhibiteur du SGLT2, actif par voie orale. En inhibant le SGLT2, la canagliflozine diminue la réabsorption du glucose filtré et diminue le seuil rénal de réabsorption du glucose (RTG). Elle augmente ainsi l'excrétion urinaire du glucose (UGE), ce qui diminue les concentrations plasmatiques du glucose par ce mécanisme indépendant de l'insuline chez les patients atteints de diabète de type 2. L'augmentation de l'UGE sous l'effet de l'inhibition du SGLT2 conduit également à une diurèse osmotique, l'effet diurétique étant lui-même à l'origine d'une diminution de la pression artérielle systolique ; l'augmentation de l'UGE se traduit par une perte de calories et donc une diminution du poids corporel, comme l'ont démontré les études effectuées chez les patients atteints de diabète de type 2.
L'action de la canagliflozine, consistant à augmenter l'UGE et à diminuer ainsi directement la glycémie, est indépendante de l'insuline. Des études cliniques avec la canagliflozine ont permis d'observer une amélioration de l'homéostasie des cellules bêta (cellules bêta HOMA) et une amélioration de la réponse insulino-sécrétoire des cellules bêta à un repas mixte.
Dans les études de phase 3, l'administration de canagliflozine 300 mg avant le repas a permis de réduire davantage l'excursion glycémique postprandiale comparativement à la dose de 100 mg. Cet effet, observé à la dose de 300 mg de canagliflozine pourrait, en partie, être dû à une inhibition locale du SGLT1 intestinal (un transporteur intestinal important du glucose) liée aux fortes concentrations transitoires de canagliflozine dans la lumière intestinale avant l'absorption du médicament (la canagliflozine est un inhibiteur peu puissant du co-transporteur SGLT1). Les études ont mis en évidence une absence de malabsorption du glucose sous canagliflozine.
La canagliflozine augmente la distribution de sodium au tubule distal en bloquant la réabsorption du sodium et du glucose dépendante de SGLT2, augmentant de ce fait le rétrocontrole tubuloglomérulaire, ce qui est associé à une réduction de la pression intraglomérulaire et à une diminution de l'hyperfiltration dans les modèles précliniques du diabète et les études cliniques.
Effets pharmacodynamiques
Après l'administration de doses orales uniques et multiples de canagliflozine chez des patients diabétiques de type 2, des diminutions dose-dépendantes du RTG et des augmentations de l'UGE ont été observées. À partir d'une valeur initiale de RTG d'environ 13 mmol/l, une suppression maximale du RTG moyen sur 24 heures a été observée avec la dose journalière à 300 mg, pour atteindre des valeurs d'environ 4 mmol/l à 5 mmol/l chez les patients diabétiques de type 2 dans les études de phase 1, ce qui suggère un faible risque d'hypoglycémie induite par le traitement. Les diminutions du RTG ont conduit à une augmentation de l'UGE chez les sujets diabétiques de type 2 traités par 100 mg ou 300 mg de canagliflozine, comprise entre 77 g/jour et 119 g/jour dans les études de phase 1 ; l'UGE observée se traduit par une perte de 308 kcal/jour à 476 kcal/jour. Les diminutions de RTG et les augmentations de l'UGE ont persisté pendant un traitement de 26 semaines chez des patients diabétiques de type 2. Des augmentations modérées (généralement < 400 mL-500 mL) du volume urinaire quotidien ont été observées ; elles se sont atténuées après plusieurs jours de traitement. La canagliflozine a augmenté de façon transitoire l'excrétion urinaire d'acide urique (augmentation de 19 % par rapport aux valeurs initiales le jour 1, avec une diminution progressive à 6 % le jour 2 et à 1 % le jour 13). Cette augmentation a été accompagnée d'une réduction prolongée de l'uricémie, d'environ 20 %.
Dans une étude à dose unique chez des patients diabétiques de type 2, l'administration de 300 mg avant un repas mixte a retardé l'absorption intestinale du glucose et a diminué la glycémie postprandiale par un mécanisme rénal mais aussi non rénal.
Efficacité et sécurité cliniques
L'amélioration du contrôle glycémique ainsi que la réduction de la morbidité et de la mortalité cardiovasculaires et rénales font partie intégrante du traitement du diabète de type 2
Efficacité glycémique et sécurité
Au total, 10 501 patients diabétiques de type 2 ont participé à dix études contrôlées en double aveugle sur l'efficacité et la sécurité clinique menées pour évaluer les effets d'Invokana sur le contrôle glycémique. La distribution ethnique était de 72 % d'origine caucasienne, 16 % d'origine asiatique, 5 % d'origine afro-américaine et 8 % d'autres. 17 % de patients étaient d'origine hispanique. 58 % des patients étaient des hommes. Les patients avaient un âge moyen de 59,5 ans (de 21 ans à 96 ans), avec 3 135 patients âgés de 65 ans et plus et 513 patients âgés de plus de 75 ans. 58 % des patients présentaient un indice de masse corporelle (IMC) ≥ 30 kg/m². Dans le programme de développement clinique, 1 085 patients avec un DFGe initial compris entre 30 mL/min/1,73 m2 et < 60 mL/min/1,73 m² ont été évalués.
Études contrôlées versus placebo
La canagliflozine a été étudiée en monothérapie, bithérapie avec la metformine, bithérapie avec un sulfamide hypoglycémiant, trithérapie avec la metformine et un sulfamide hypoglycémiant, trithérapie avec la metformine et la pioglitazone et en association à l'insuline (tableau 4). En général, la canagliflozine a produit des résultats cliniquement et statistiquement significatifs (p < 0,001) par rapport au placebo concernant le contrôle glycémique, y compris le taux d'HbA1c, le pourcentage de patients atteignant un taux d'HbA1c < 7 %, la modification de la glycémie à jeun par rapport aux valeurs initiales et la glycémie postprandiale à 2 heures. En outre, des diminutions du poids corporel et de la pression artérielle systolique par rapport au placebo ont été observées.
Par ailleurs, la canagliflozine a été étudiée en trithérapie avec la metformine et la sitagliptine et dosée avec un schéma de progression posologique utilisant une dose de départ de 100 mg augmentée à 300 mg dès la semaine 6 chez les patients ayant besoin d'un contrôle glycémique additionnel, ayant eu un taux de filtration glomérulaire estimé approprié, et ayant toléré la canagliflozine dosée à 100 mg (tableau 4). La canagliflozine dosée avec un schéma de progression posologique a produit des résultats cliniquement et statistiquement significatifs (p < 0,001) par rapport au placebo en termes de contrôle glycémique, incluant le taux d'HbA1c et la variation du glucose plasmatique à jeun par rapport à la valeur initiale, ainsi qu'une amélioration statistiquement significative (p < 0,01) du pourcentage de patients atteignant un taux d'HbA1c < 7 %. De plus, par rapport au placebo, des diminutions du poids corporel et de la pression artérielle systolique ont été observées.
Tableau 4 : Résultats d'efficacité des études cliniques contrôlées versus placeboa
| Monothérapie (26 semaines) | |||
| Canagliflozine | Placebo (N = 192) | ||
| 100 mg (N = 195) | 300 mg (N = 197) | ||
| HbA1c (%) | |||
| Valeur initiale (moyenne) | 8,06 | 8,01 | 7,97 |
| Variation par rapport aux valeurs initiales (moyenne ajustée) | -0,77 | -1,03 | 0,14 |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -0,91b(-1,09 ; - 0,73) | -1,16b(-1,34 ; -0,98) | Sans objetc |
| Patients (%) atteignant un taux d'HbA1c < 7 % | 44,5b | 62,4b | 20,6 |
| Poids corporel | |||
| Valeur initiale (moyenne) en kg | 85,9 | 86,9 | 87,5 |
| Pourcentage de variation par rapport aux valeurs initiales (moyenne ajustée) | -2,8 | -3,9 | -0,6 |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -2,2b(-2,9 ; -1,6) | -3,3b(-4,0 ; -2,6) | Sans objetc |
| Bithérapie avec la metformine (26 semaines) | |||
| Canagliflozine + metformine | Placebo
+ metformine (N = 183) |
||
| 100 mg (N = 368) | 300 mg (N = 367) | ||
| HbA1c (%) | |||
| Valeur initiale (moyenne) | 7,94 | 7,95 | 7,96 |
| Variation par rapport aux valeurs initiales (moyenne ajustée) | -0,79 | -0,94 | -0,17 |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -0,62b(-0,76 ; - 0,48) | -0,77b(-0,91 ; -0,64) | Sans objetc |
| Patients (%) atteignant un taux d'HbA1c < 7 % | 45,5b | 57,8b | 29,8 |
| Poids corporel | |||
| Valeur initiale (moyenne) en kg | 88,7 | 85,4 | 86,7 |
| Pourcentage de variation par rapport aux valeurs initiales (moyenne ajustée) | -3,7 | -4,2 | -1,2 |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -2,5b(-3,1 ; -1,9) | -2,9b(-3,5 ; -2,3) | Sans objetc |
| Trithérapie avec la metformine et un sulfamide hypoglycémiant (26 semaines) | |||
| Canagliflozine + metformine et sulfamide hypoglycémiant | Placebo
+ metformine et sulfamide hypoglycémiant (N = 156) |
||
| 100 mg (N = 157) | 300 mg (N = 156) | ||
| HbA1c (%) | |||
| Valeur initiale (moyenne) | 8,13 | 8,13 | 8,12 |
| Variation par rapport aux valeurs initiales (moyenne ajustée) | -0,85 | -1,06 | -0,13 |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -0,71b(-0,90 ;-0,52) | -0,92b(-1,11 ;-0,73) | Sans objetc |
| Patients (%) atteignant un taux d'HbA1c < 7 % | 43,2b | 56,6b | 18,0 |
| Poids corporel | |||
| Valeur initiale (moyenne) en kg | 93,5 | 93,5 | 90,8 |
| Pourcentage de variation par rapport aux valeurs initiales (moyenne ajustée) | -2,1 |
-2,6 | -0,7 |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -1,4b(-2,1 ;-0,7) | -2,0b(-2,7 ;-1,3) | Sans objetc |
| En association à l'insulined (18 semaines) | |||
| Canagliflozine + insuline | Placebo + insuline (N = 565) | ||
| 100 mg (N = 566) | 300 mg (N = 587) | ||
| HbA1c (%) | |||
| Valeur initiale (moyenne) | 8,33 | 8,27 | 8,20 |
| Variation par rapport aux valeurs initiales (moyenne ajustée) | -0,63 | -0,72 | 0,01 |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -0,65b(-0,73 ; -0,56) | -0,73b(-0,82 ; -0,65) | Sans objetc |
| Patients (%) atteignant un taux d'HbA1c < 7 % | 19,8b | 24,7b | 7,7 |
| Poids corporel | |||
| Valeur initiale (moyenne) en kg | 96,9 | 96,7 | 97,7 |
| Pourcentage de variation par rapport aux valeurs initiales (moyenne ajustée) | -1,8 | -2,3 | 0,1 |
| Différence par rapport au placebo (moyenne ajustée) (IC 97,5 %) | -1,9b(-2,2 ; -1,5) | -2,4b(-2,8 ; -2,0) | Sans objetc |
| Trithérapie avec la metformine et la sitagliptinee (26 semaines) | |||
| Canagliflozine + metformine et sitagliptineg (N = 107) | Placebo + metformine et sitagliptine (N = 106) | ||
| HbA1c (%) | |||
| Valeur initiale (moyenne) | 8,53 | 8,38 | |
| Variation par rapport aux valeurs initiales (moyenne ajustée) | -0,91 | -0,01 | |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -0,89b(-1,19 ; -0,59) | ||
| Patients (%) atteignant un taux d'HbA1c < 7 % | 32f | 12 | |
| Glucose plasmatique à jeun (mg/dL) | |||
| Valeur initiale (moyenne) | 186 | 180 | |
| Variation par rapport aux valeurs initiales (moyenneajustée) | -30 | -3 | |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -27b(-40 ; -14) | ||
| Poids corporel | |||
| Valeur initiale (moyenne) en kg | 93,8 | 89,9 | |
| Variation par rapport aux valeurs initiales en % (moyenne ajustée) | -3,4 | -1,6 | |
| Différence par rapport au placebo (moyenne ajustée) (IC 95 %) | -1,8b(-2,7 ; -0,9) | ||
b p < 0,001 comparativement au placebo.
c Non applicable.
d canagliflozine en association à l'insuline (avec ou sans autres médicaments hypoglycémiants).
e canagliflozine 100 mg augmentée à 300 mg
f p < 0,01 par rapport au placebo
g 90,7 % des sujets du groupe canagliflozine avec dose augmentée à 300 mg
Outre les études ci-dessus, les résultats d'efficacité observés sur la glycémie dans une sous-étude de bithérapie de 18 semaines avec un sulfamide hypoglycémiant et une étude de trithérapie de 26 semaines avec la metformine et la pioglitazone ont été généralement comparables à ceux observés dans d'autres études.
Études contrôlées versus comparateur actif
La canagliflozine a été comparée au glimépiride en bithérapie avec la metformine et à la sitagliptine en trithérapie avec la metformine et un sulfamide hypoglycémiant (tableau 5). Canagliflozine 100 mg en bithérapie avec la metformine a entrainé des diminutions similaires de taux d'HbA1c par rapport aux valeurs initiales et à une dose de 300 mg a entrainé des diminutions supérieures (p < 0,05) de l'HbA1c, comparativement au glimépiride, ce qui démontre la non-infériorité. Une proportion plus faible de patients traités par canagliflozine 100 mg (5,6 %) et canagliflozine 300 mg (4,9 %) a présenté au moins un épisode/événement d'hypoglycémie au cours des 52 semaines de traitement, comparativement au groupe traité par glimépiride (34,2 %). Dans une étude comparant la canagliflozine 300 mg à la sitagliptine 100 mg en trithérapie avec la metformine et un sulfamide hypoglycémiant, la canagliflozine a été à l'origine de diminutions non inférieures (p < 0,05) et supérieures (p < 0,05) du taux d'HbA1c par rapport à la sitagliptine. L'incidence des épisodes/événements d'hypoglycémies sous canagliflozine 300 mg et sitagliptine 100 mg a été respectivement de 40,7 % and 43,2 %. Des améliorations significatives du poids corporel et des diminutions significatives de la pression artérielle systolique ont également été observées comparativement au glimépiride et à la sitagliptine.
Tableau 5 : Résultats d'efficacité des études cliniques contrôlées versus comparateur actifa
| Comparaison au glimépiride en bithérapie avec la metformine (52 semaines) | |||||
| Canagliflozine + metformine | Glimépiride (titré) + metformine (N = 482) | ||||
| 100
mg (N = 483) |
300
mg (N = 485) |
||||
| HbA1c (%) | |||||
| Valeur initiale (moyenne) | 7,78 | 7,79 | 7,83 | ||
| Variation par rapport aux valeurs initiales (moyenne ajustée) | -0,82 | -0,93 | -0,81 | ||
| Différence par rapport au glimépiride (moyenne ajustée) (IC 95 %) | -0,01b(-0,11 ; 0,09) | -0,12b (-0,22 ; -0,02) | Sans objetc | ||
| Patients (%) atteignant un taux d'HbA1c< 7 % | 53,6 | 60,1 | 55,8 | ||
| Poids corporel | |||||
| Valeur initiale (moyenne) en kg | 86,8 | 86,6 | 86,6 | ||
| Pourcentage de variation par rapport aux valeurs initiales (moyenne ajustée) | -4,2 | -4,7 | 1,0 | ||
| Différence par rapport au glimépiride (moyenne ajustée) (IC 95 %) | -5,2b(-5,7 ; -4,7) | -5,7b(-6,2 ; -5,1) | Sans objetc | ||
| Comparaison à la sitagliptine en trithérapie avec la metformine et un sulfamide hypoglycémiant (52 semaines) | |||||
| Canagliflozine 300 mg + metformine et sulfamide hypoglycémiant (N = 377) | Sitagliptine 100 mg+ metformine et sulfamide hypoglycémiant (N = 378) | ||||
| HbA1c (%) | |||||
| Valeur initiale (moyenne) | 8,12 | 8,13 | |||
| Variation par rapport aux valeurs initiales (moyenne ajustée) | -1,03 | -0,66 | |||
| Différence par rapport à la sitagliptine (moyenne ajustée) (IC 95 %) | -0,37b(-0,50 ; -0,25) | NAc | |||
| Patients (%) atteignant un taux d'HbA1c< 7 % | 47,6 | 35,3 | |||
| Poids corporel | |||||
| Valeur initiale (moyenne) en kg | 87,6 | 89,6 | |||
| Pourcentage de variation par rapport aux valeurs initiales (moyenne ajustée) | -2,5 | 0,3 | |||
| Différence par rapport à la sitagliptine (moyenne ajustée) (IC 95 %) | -2,8d(-3,3 ; -2,2) | Sans objetc | |||
a Population en intention de traiter
utilisant la dernière observation de l'étude avant le recours à un
traitement de secours.
b p < 0,05.
c Non applicable.
d p < 0,001.
b p < 0,05.
c Non applicable.
d p < 0,001.
Canagliflozine en association avec la metformine en traitement initial
La canagliflozine a été évaluée en association avec la metformine comme traitement initial chez les patients avec un diabète de type 2 chez qui la perte de poids et l'exercice physique ont échoué. La canagliflozine dosée à 100 mg et la canagliflozine dosée à 300 mg en association avec la metformine XR a résulté en une amélioration statistiquement significative du taux d'HbA1C par rapport aux doses respectives de canagliflozine (100 mg et 300 mg) seule ou de metformine XR seule (tableau 6).
Tableau 6 : Résultats d'un essai clinique de 26 semaines contrôlé avec comparateur actif étudiant la canagliflozine en association avec la metformine en traitement initial *
| Paramètre d'efficacité | Metformine
XR (N = 237) |
Canagliflozine
100 mg (N = 237) |
Canagliflozine
300 mg (N = 238) |
Canagliflozine
100 mg + Metformine XR (N = 237) |
Canagliflozine
300 mg + Metformine XR (N = 237) |
| HbA1c (%) | |||||
| Valeur initiale (moyenne) | 8,81 | 8,78 | 8,77 | 8,83 | 8,90 |
| Variation par
rapport aux valeurs initiales (moyenne ajustée) |
-1,30 | -1,37 | -1,42 | -1,77 | -1,78 |
| Différence
par rapport à la canagliflozine 100 mg (moyenne ajustée) (IC95 %) † |
-0,40‡(-0,59, -0,21) | ||||
| Différence
par rapport à la canagliflozine 300 mg (moyenne ajustée) (IC 95 %) † |
-0,36‡(-0,56, - 0,17) | ||||
| Différence
par rapport à la metformine XR (moyenne ajustée) (IC95 %) † |
-0,06‡(-0,26, 0,13) | -0,11‡(-0,31, 0,08) | -0,46‡(-0,66, -0,27) | -0,48‡(-0,67, - 0,28) | |
| Patients
(%) atteignant un taux d'HbA1c< 7 % |
43 | 39 | 43 | 50§§ | 57§§ |
| Poids corporel | |||||
| Valeur initiale (moyenne) en kg | 92,1 | 90,3 | 93,0 | 88,3 | 91,5 |
| Pourcentage
de variation par rapport aux valeurs initiales (moyenne ajustée) |
-2,1 | -3,0 | -3,9 | -3,5 | -4,2 |
| Différence
par rapport à la metformine XR (moyenne ajustée) (IC 95 %) † |
-0,9§§(-1,6, -0,2) | -1,8§(-2,6, -1,1) | -1,4‡(-2,1, -0,6) | -2,1‡(-2,9, -1,4) | |
* Population en intention
de traiter
† Moyenne ajustée des moindres carrés pour les covariables incluant la valeur initiale et le facteur de stratification
‡ p = 0,001 ajusté
§ p < 0,01 ajusté
§§ p < 0,05 ajusté
† Moyenne ajustée des moindres carrés pour les covariables incluant la valeur initiale et le facteur de stratification
‡ p = 0,001 ajusté
§ p < 0,01 ajusté
§§ p < 0,05 ajusté
Populations particulières
Dans trois études menées dans des populations particulières (patients âgés, patients avec un DFGe ≥ 30 mL/min/1,73 m2 et < 50 mL/min/1,73 m² et patients présentant un risque élevé de maladie cardiovasculaire ou une maladie cardiovasculaire avérée), la canagliflozine a été ajoutée chez des patients équilibrés par traitements antidiabétiques (régime alimentaire, monothérapie ou association thérapeutique).
Patients âgés
Au total, 714 patients âgés de ≥ 55 ans à ≤ 80 ans (227 patients âgés de 65 ans à < 75 ans et 46 patients âgés de 75 ans à ≤ 80 ans) ayant un contrôle glycémique insuffisant avec leur traitement antidiabétique en cours (médicaments hypoglycémiants et/ou régime alimentaire et pratique d'une activité physique) ont participé à une étude en double aveugle, contrôlée versus placebo pendant 26 semaines. Des modifications statistiquement significatives (p < 0,001) par rapport aux valeurs initiales d'HbA1c comparativement au placebo, atteignant -0,57 % et -0,70 %, ont été observées pour la dose de 100 mg et celle de 300 mg respectivement (voir rubriques Posologie et mode d'administration et Effets indésirables).
Patients avec un DFGe < 60 mL/min/1,73 m²
Dans une analyse poolée des patients (N = 721) avec un DFGe initial ≥ 45 mL/min/1,73 m2 et < 60 mL/min/1,73 m², la canagliflozine a été à l'origine d'une diminution cliniquement significative du taux d'HbA1c comparativement au placebo, avec des valeurs atteignant -0,47 % pour 100 mg de canagliflozine et -0,52 % pour 300 mg de canagliflozine. Les patients avec un DFGe initial ≥ 45 mL/min/1,73 m2 et < 60 mL/min/1,73 m² traités par la canagliflozine 100 mg et 300 mg ont présenté des améliorations moyennes du poids corporel de respectivement -1,8 % et -2,0 % par rapport au placebo
Dans une analyse poolée de patients (N = 348) dont le DFGe initial était < 45 mL/min/1.73 m², la canagliflozine a entraîné une baisse modeste du taux d'HbA1c comparativement au placebo, avec -0,23 % pour la canagliflozine à 100 mg et -0,39 % pour la canagliflozine à 300 mg.
La majorité des patients avec un DFGe initial < 60 mL/min/1,73 m² étaient traités par insuline et/ou sulfamide hypoglycémiant. Comme attendu lorsqu'un produit non associé à des hypoglycémies est ajouté à l'insuline et/ou à un sulfamide hypoglycémiant, une augmentation des épisodes/événements d'hypoglycémies a été observée lorsque la canagliflozine a été associée à l'insuline et/ou à un sulfamide hypoglycémiant (voir rubrique Effets indésirables).
Glycémie à jeun
Dans quatre études contrôlées versus placebo, le traitement par canagliflozine en monothérapie ou en association avec un ou deux médicaments hypoglycémiants oraux a provoqué des modifications moyennes de la glycémie à jeun, par rapport aux valeurs initiales, de -1,2 mmol/l à -1,9 mmol/l pour canagliflozine 100 mg et de -1,9 mmol/l à -2,4 mmol/l pour canagliflozine 300 mg, comparativement au placebo. Ces diminutions ont été maintenues pendant toute la période de traitement et étaient presque maximales dès le premier jour de traitement.
Glycémie postprandiale
Lors d'une épreuve d'exposition à un repas mixte, canagliflozine en monothérapie ou en association avec un ou deux médicaments hypoglycémiants oraux a diminué la glycémie postprandiale, par rapport aux valeurs initiales, de respectivement -1,5 mmol/l à - 2,7 mmol/l pour canagliflozine 100 mg et de -2,1 mmol/l à -3,5 mmol/l pour canagliflozine 300 mg, comparativement au placebo, en raison des diminutions des glycémies préprandiales et de la diminution des excursions glycémiques postprandiales.
Poids corporel
Canagliflozine 100 mg et 300 mg en monothérapie et en association en bithérapie ou trithérapie a été à l'origine de diminutions statistiquement significatives du poids corporel à 26 semaines, comparativement au placebo. Dans deux études contrôlées versus comparateur actif de 52 semaines, comparant la canagliflozine au glimépiride et à la sitagliptine, il a été observé des diminutions moyennes prolongées et statistiquement significatives du poids corporel de respectivement - 4,2 % et - 4,7 % pour canagliflozine 100 mg et 300 mg associés à la metformine, comparativement à l'association de glimépiride et metformine (1,0 %). Cette diminution a été de - 2,5 % pour canagliflozine 300 mg en association à la metformine et un sulfamide hypoglycémiant,comparativement à la sitagliptine en association à la metformine et un sulfamide hypoglycémiant (0,3 %).
Un sous-ensemble de patients (N = 208) de l'étude en bithérapie avec la metformine, contrôlée versus comparateur actif, a subi des examens par absorptiométrie à rayons X double énergie (DXA) et une tomodensitométrie abdominale pour évaluer la composition corporelle. Il a ainsi été démontré qu'environ deux tiers de la perte de poids sous canagliflozine étaient dus à une perte de masse grasse, avec des quantités similaires de pertes de graisse viscérale et sous-cutanée abdominale. Deux cent onze (211) patients de l'étude clinique menée chez des patients âgés ont participé à une sous-étude évaluant la composition corporelle au moyen d'une analyse DXA. Cette étude a démontré qu'environ deux tiers de la perte de poids associée à la canagliflozine étaient dus à la perte de masse grasse, comparativement au placebo. Aucune modification significative de la densité osseuse dans les régions trabéculaires et corticales n'a été mise en évidence.
Pression artérielle
Lors des études controlées contre placebo, le traitement par canagliflozine 100 mg et 300 mg a donné lieu à des réductions moyennes de la pression artérielle systolique de, respectivement, -3,9 mmHg et -5,3 mmHg, par rapport au placebo (-0,1 mmHg) et à un moindre effet sur la pression artérielle diastolique avec des modifications moyennes pour canagliflozine 100 mg et 300 mg de, respectivement, -2,1 mmHg et -2,5 mmHg, par rapport au placebo (-0,3 mmHg). Il n'y a pas eu de modification notable de la fréquence cardiaque.
Patients présentant un taux d'HbA1c de référence > 10% et ≤ 12%
Une sous-étude menée auprès de patients dont le taux d'HbA1c de référence est > 10% et ≤ 12% avec la canagliflozine en monothérapie a mis en évidence des réductions par rapport à la valeur d'HbA1c de référence (non ajustée par rapport au placebo) respectivement de -2,13 % et -2,56 % pour canagliflozine 100 mg et 300 mg.
Résultats cardiovasculaires dans le programme CANVAS
L'effet de la canagliflozine sur les événements cardiovasculaires chez l'adulte souffrant de diabète de type 2 et présentant une maladie cardiovasculaire (CV) établie ou un risque de maladie cardiovasculaire (deux facteurs de risque cardiovasculaires ou plus), a été évalué dans le cadre du programme CANVAS (analyse intégrée des études CANVAS et CANVAS-R). Ces études étaient multicentriques, internationales, randomisées, en double-aveugle, en groupes parallèles, avec des critères d'inclusion et d'exclusion ainsi que des populations de patients similaires. Le programme CANVAS comparait le risque de survenue d'un événement cardiovasculaire indésirable majeur (ECIM), défini comme étant composé d'une mort cardiovasculaire, d'un infarctus du myocarde non mortel et d'un accident vasculaire cérébral non mortel, entre la canagliflozine et le placebo, dans un contexte de traitement standard du diabète et de la maladie cardiovasculaire athérosclérotique.
Dans l'essai CANVAS, les sujets ont été randomisés selon un rapport de 1:1:1 vers la canagliflozine 100 mg, la canagliflozine 300 mg ou un placebo correspondant. Dans l'essai CANVAS-R, les sujets ont été randomisés selon un rapport de 1:1 vers la canagliflozine 100 mg ou un placebo correspondant et une titration à 300 mg a été autorisée (en fonction de la tolérance et des besoins glycémiques) après la Semaine 13. Les traitements antidiabétiques et de l'athérosclérose concomitants ont pu être ajustés, selon la prise en charge standard de ces maladies.
Au total, 10 134 patients ont été traités (4 327 dans l'essai CANVAS et 5 807 dans l'essai CANVAS- R ; un total de 4 344 patients a été randomisé dans le groupe placebo et 5 790 dans le groupe canagliflozine) pendant une durée moyenne d'exposition de 149 semaines (223 semaines dans l'essai CANVAS et 94 semaines dans l'essai CANVAS-R). Le statut vital a été obtenu pour 99,6 % des sujets sur l'ensemble des études. L'âge moyen était de 63 ans et 64 % étaient des hommes. Soixante-six pourcent des sujets avaient un antécédent de maladie cardiovasculaire établie, 56 % ayant souffert d'une coronaropathie, 19 % d'une maladie vasculaire cérébrale et 21 % d'une maladie vasculaire périphérique ; 14 % avaient un antécédent d'insuffisance cardiaque.
Le taux moyen d'HbA1c de référence était de 8,2 % et la durée moyenne du diabète était de 13,5 ans.
Les patients devaient présenter un DFGe > 30 mL/min/1,73 m² au début de l'étude. La fonction rénale de référence était normale ou légèrement altérée chez 80 % des patients et modérément altérée chez 20 % des patients (DFGe moyen de 77 ml/min/1,73 m2). A l'origine, les patients recevaient un ou plusieurs médicaments antidiabétiques tels que de la metformine (77 %), de l'insuline (50 %) et des sulfonylurées (43 %).
Le principal critère d'évaluation du programme CANVAS était le temps jusqu'à la première survenue d'un ECIM. Les critères secondaires d'une vérification d'hypothèses conditionnelles séquentielles étaient la mortalité toutes causes confondues et la mortalité cardiovasculaire.
Les patients inclus dans les groupes canagliflozine poolés (analyse poolée portant sur canagliflozine 100 mg, canagliflozine 300 mg et canagliflozine dont le dosage passe de 100 mg à 300 mg) présentaient un taux d'ECIM inférieur par rapport au placebo : 2,69 versus 3,15 patients pour 100 patients-années (RR de l'analyse poolée : 0,86 ; IC 95 % (0,75, 0,97).
Selon la courbe de Kaplan-Meier relative à la première survenue d'un ECIM, présentée ci-dessous, la réduction des ECIM au sein du groupe canagliflozine a été observée dés la semaine 26 et s'est maintenue jusqu'à la fin de l'étude (voir Figure 1).
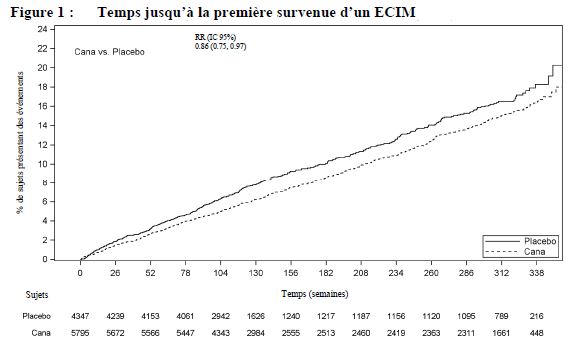
2 011 patients présentaient un DFGe compris entre 30 et < 60 ml/min/1,73 m2. Les résultats concernant les ECIM dans les sous-groupes entre 30 et < 60 mL/min/1,73 m², entre 30 et < 45 mL/min/1,73 m² et entre 45 et < 60 mL/min/1,73 m² étaient cohérents avec les résultats généraux.
Chaque ECIM a positivement contribué au résultat global tel qu'indiqué par la Figure 2. Les résulats relatifs aux doses de 100 mg et 300 mg de canagliflozine étaient cohérents avec ceux des groupes relatifs aux doses combinés.
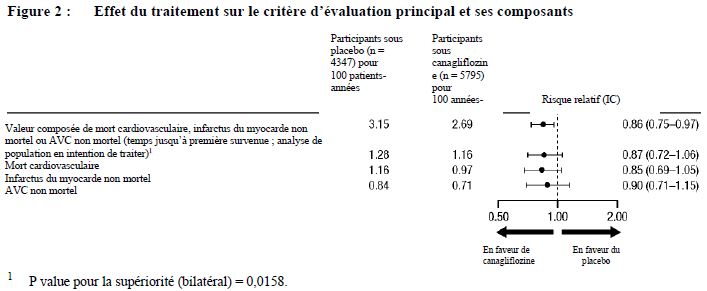
Mortalité toutes causes confondues dans le programme CANVAS
Au sein du groupe canagliflozine combiné, le RR pour la mortalité toutes causes confondues par rapport au placebo était de 0,87 ; IC 95 % (0,74, 1,01).
Insuffisance cardiaque nécessitant une hospitalisation dans le programme CANVAS
La canagliflozine a réduit le risque d'insuffisance cardiaque nécessitant une hospitalisation par rapport au placebo (RR : 0,67 ; IC 95 % (0,52, 0,87)).
Critères d'évaluation rénaux dans le programme CANVAS
Concernant la survenue d'un premier évènement évoquant une néphropathie (doublement de la créatinine sérique, nécessité d'une thérapie de remplacement rénal, et mort rénale), le RR était de 0,53 (95% IC : 0,33 ; 0,84) pour la canagliflozine (0,15 évènement pour 100 patients-années) versus placebo (0,28 évènement pour 100 patients-années). De plus, la canagliflozine réduit la progression de l'albuminurie de 25,8% versus placebo 29,2% (RR : 0,73 ; 95% IC : 0,67 ; 0,79) chez les patients ayant une valeur de référence correspondant à une normo ou une micro-albuminurie.
Devenir de la fonction rénale dans l'étude CREDENCE
L'effet de la canagliflozine 100 mg sur les événements rénaux chez des adultes atteints de diabète de type 2 et de maladie rénale chronique (MRC) ayant un débit de filtration glomérulaire estimé (DFGe) de 30 à < 90 mL/min/1,73 m2 et une albuminurie ( > 300 à 5 000 mg/g de créatinine) a été évalué dans l'étude CREDENCE (Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation Trial). Cette étude était multicentrique, internationale, randomisée, en double- aveugle, axée sur les événements, contrôlée contre placebo, en groupes parallèles. L'étude CREDENCE comparait le risque de survenue d'une MRC définie comme étant composée d'une insuffisance rénale terminale (IRT), du doublement de la créatinine sérique, et de la mort cardiovasculaire ou rénale, entre la canagliflozine 100 mg et le placebo dans un contexte de traitement standard pour la MRC, y compris un inhibiteur de l'enzyme de conversion de l'angiotensine (IEC) ou un antagoniste des récepteurs de l'angiotensine (ARA II). La canagliflozine 300 mg n'a pas été évaluée dans cette étude.
Dans l'étude CREDENCE, les sujets ont été randomisés selon un rapport de 1:1 à recevoir la canagliflozine 100 mg ou le placebo, puis stratifiés en fonction du DFGe de 30 à < 45, de 45 à < 60, ou de 60 à < 90 mL/min/1,73 m2. Le traitement par canagliflozine 100 mg a été poursuivi chez les patients jusqu'à l'initiation de la dialyse ou jusqu'à une transplantation rénale.
Au total, 4 397 sujets ont été traités et exposés pendant une durée moyenne de 115 semaines. L'âge moyen des sujets était de 63 ans, et 66 % étaient des hommes.
Le taux d'HbA1c moyen de référence était de 8,3 % et l'albumine/créatinine urinaire médiane initiale était de 927 mg/g. Les agents anti-hyperglycémiques (AAH) les plus fréquemment utilisés à l'inclusion étaient l'insuline (65,5 %), des biguanides (57,8 %) et des sulfamides hypoglycémiants (28,8 %). Presque tous les sujets (99,9 %) étaient sous traitement par IEC et ARA II à la randomisation. Près de 92 % des sujets étaient sous traitements cardiovasculaires (sans compter les traitements par IEC/ARA II) à l'inclusion, environ 60 % prenaient un agent antithrombotique (notamment de l'acide acétylsalicylique) et 69 % étaient sous statines.
Le DFGe moyen de référence était de 56,2 mL/min/1,73 m2 et environ 60 % de la population avait un DFGe de référence < 60 mL/min/1,73 m2. La proportion de sujets ayant des antécédents de maladie CV était de 50,4 % ; 14,8 % avaient des antécédents d'insuffisance cardiaque.
Le critère d'évaluation composite principal de l'étude CREDENCE était le temps jusqu'à la première survenue d'une IRT (définie comme un DFGe < 15 mL/min/1,73 m2, l'initiation d'une dialyse chronique ou une transplantion rénale), du doublement de la créatinine sérique, et de la mort CV ou rénale.
La canagliflozine 100 mg a réduit significativement le risque de première survenue du critère d'évaluation composite principal d'IRT, de doublement de la créatinine sérique, et de mort CV ou rénale [p< 0,0001 ; RR : 0,70 ; IC 95 % : 0,57, 0,84] (voir figure 4). L'effet du traitement était cohérent dans tous les sous-groupes, y compris les trois strates de DFGe et les sujets avec ou sans antécédents de maladie CV.
Selon la courbe de Kaplan-Meier du temps jusqu'à la première survenue du critère d'évaluation composite principal, présentée ci-dessous, l'effet du traitement était évident à partir de la semaine 52 avec la canagliflozine 100 mg et était maintenu jusqu'à la fin de l'étude (voir figure 3).
La canagliflozine 100 mg réduisait significativement le risque de survenue des critères d'évaluation secondaires cardiovasculaires, comme indiqué en figure 4.
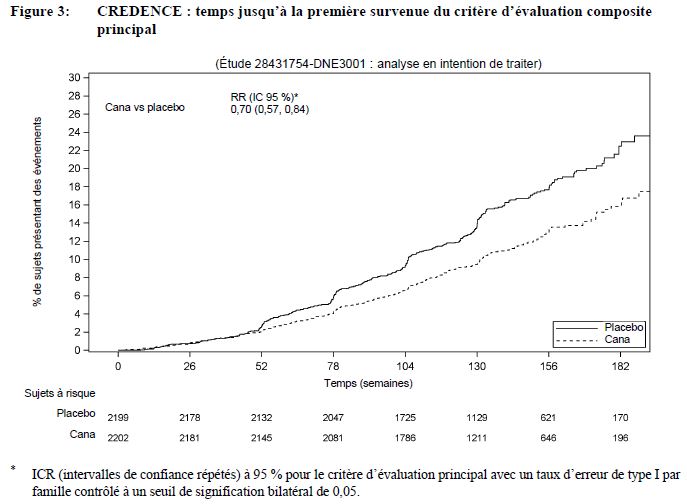
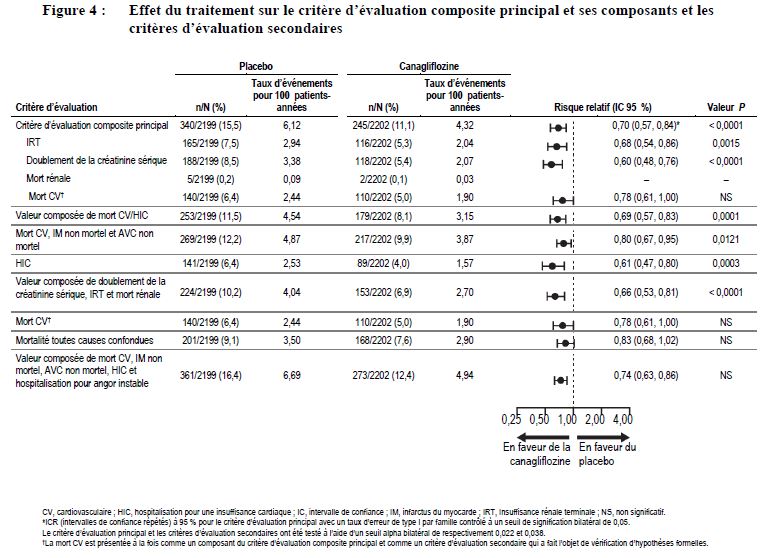
Comme l'illustre la figure 5, le DFGe des patients traité par placebo montrait une diminution linéaire progressive avec le temps ; en revanche, le groupe canagliflozine montrait une baisse aiguë à la semaine 3, suivie par une diminution atténuée avec le temps ; après la semaine 52, la diminution moyenne des moindres carrés du DFGe était plus faible dans le groupe canagliflozine que dans le groupe placebo, et l'effet du traitement était maintenu jusqu'à la fin du traitement.
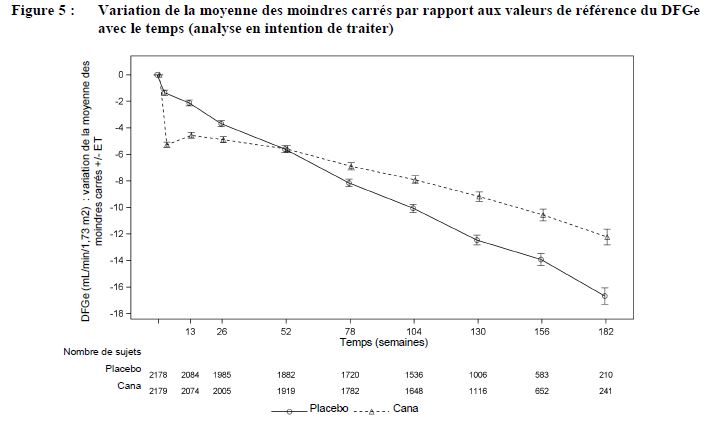
Dans l'étude CREDENCE, le taux d'incidence des événements indésirables liés à la fonction rénale était inférieur dans le groupe traité par canagliflozine 100 mg par rapport au groupe placebo (5,71 et 7,91 pour 100 patients-années respectivement avec la canagliflozine 100 mg et le placebo).
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec la canagliflozine dans un ou plusieurs sous-ensembles de la population pédiatrique atteinte d'un diabète de type 2 (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La
pharmacocinétique de la canagliflozine est similaire chez les sujets
sains et les patients diabétiques de type 2. Après l'administration
d'une dose orale unique de 100 mg ou 300 mg chez des sujets sains, la
canagliflozine a été rapidement absorbée, avec des concentrations
plasmatiques maximales apparaissant 1 heure à 2 heures après l'administration de la dose (Tmax médian). La Cmax plasmatique et l'ASC de la canagliflozine ont augmenté de manière dose-dépendante de 50 mg à 300 mg. La demi-vie apparente (t1/2) (exprimée en moyenne ± écart-type) a été respectivement de 10,6 ± 2,13 heures et de 13,1 ± 3,28 heures pour les doses de 100 mg et 300 mg. L'équilibre a été atteint après 4 jours à 5
jours de traitement par canagliflozine aux doses de 100 mg à 300 mg une
fois par jour. La canagliflozine ne présente pas un profil de
pharmacocinétique temps-dépendant et s'est accumulée dans le plasma
jusqu'à 36 % après des doses multiples de 100 mg et 300 mg.
Absorption
La biodisponibilité orale absolue moyenne de la canagliflozine est d'environ 65 %. La co- administration d'un repas riche en lipides avec la canagliflozine n'a pas eu d'effet sur la pharmacocinétique de la canagliflozine ; par conséquent, Invokana peut être pris avec ou sans aliments. Toutefois, en raison de la diminution potentielle des excursions glycémiques postprandiales liées au retard d'absorption intestinale du glucose, il est recommandé de prendre Invokana avant le premier repas de la journée (voir rubriques Posologie et mode d'administration et Propriétés pharmacodynamiques).
Distribution
Le volume moyen de distribution de la canagliflozine à l'équilibre, après une seule perfusion intraveineuse chez des sujets sains, a été de 83,5 litres, ce qui suggère une distribution tissulaire importante. La canagliflozine est fortement liée aux protéines dans le plasma (99 %), principalement à l'albumine. La liaison aux protéines est indépendante des concentrations plasmatiques de la canagliflozine. La liaison aux protéines plasmatiques n'est pas modifiée de façon significative chez les patients atteints d'insuffisance rénale ou hépatique.
Biotransformation
La réaction d'O-glucuronidation est la voie d'élimination métabolique principale de la canagliflozine, qui est essentiellement transformée par les enzymes UGT1A9 et UGT2B4 en deux métabolites O- glucuronides inactifs. Le métabolisme (oxydatif) de la canagliflozine médié par CYP3A4 est minime (environ 7 %) chez l'homme.
Dans les études in vitro, la canagliflozine n'a pas inhibé les isoenzymes du cytochrome P450 CYP1A2, CYP2A6, CYP2C19, CYP2D6, ou CYP2E1, CYP2B6, CYP2C8, CYP2C9, ni induit les CYP1A2, CYP2C19, CYP2B6, CYP3A4 lorsqu'elle était utilisée à des concentrations supérieures aux concentrations thérapeutiques. Aucun effet cliniquement pertinent sur le CYP3A4 n'a été observé in vivo (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions)
Élimination
Après l'administration d'une dose orale unique de [C14]canagliflozine chez des sujets sains, 41,5 %, 7,0 % et 3,2 % de la dose radioactive administrée ont été retrouvés dans les fèces respectivement sous forme de canagliflozine, de métabolites hydroxylés et de métabolites O-glucuronide. La circulation entérohépatique de la canagliflozine a été négligeable.
Environ 33 % de la dose radioactive administrée a été excrétée dans l'urine, principalement sous forme de métabolites O-glucuronides (30,5 %). Moins de 1 % de la dose a été excrété sous forme de canagliflozine inchangée dans l'urine. La clairance rénale de la canagliflozine pour les doses à 100 mg et 300 mg était comprise entre 1,30 mL/min et 1,55 mL/min.
La canagliflozine est une substance à faible clairance, avec une clairance systémique moyenne d'environ 192 mL/min chez les sujets sains, après une administration intraveineuse.
Populations particulières
Insuffisance rénale
Une étude ouverte, à dose unique, a évalué la pharmacocinétique de la canagliflozine à 200 mg chez des sujets ayant différents degrés d'insuffisance rénale (classés selon la clairance de la créatinine (ClCr) d'après l'équation de Cockroft-Gault), comparativement à des sujets sains. L'étude a inclus 8 sujets ayant une fonction rénale normale (ClCr ≥ 80 mL/min), 8 sujets ayant une insuffisance rénale légère (ClCr ≥ 50 mL/min et < 80 mL/min), 8 sujets ayant une insuffisance rénale modérée(ClCr ≥ 30 mL/min et < 50 mL/min) et 8 sujets ayant une insuffisance rénale sévère (ClCr < 30 mL/min), ainsi que 8 sujets en insuffisance rénale terminale (IRT) sous hémodialyse.
La Cmax de la canagliflozine a été modérément augmentée respectivement de 13 %, 29 % et 29 % chez les sujets présentant une insuffisance rénale légère, modérée et sévère, mais pas chez les sujets sous hémodialyse. Comparativement aux sujets sains, l'ASC plasmatique de la canagliflozine a été augmentée respectivement d'environ 17 %, 63 % et 50 % chez les sujets ayant une insuffisance rénale légère, modérée et sévère, mais a été similaire chez les sujets en IRT et les sujets sains.
La canagliflozine a été éliminée de façon négligeable par hémodialyse.
Insuffisance hépatique
Par rapport aux sujets ayant une fonction hépatique normale, les rapports des moyennes géométriques pour les Cmax et les ASC∞ de la canagliflozine ont été respectivement de 107 % et 110 %, chez les sujets atteints d'une insuffisance hépatique légère (classe A de Child-Pugh) et respectivement de 96 % et 111 %, chez les sujets atteints d'une insuffisance hépatique de classe B de Child-Pugh (modérée) après l'administration d'une dose unique de 300 mg de canagliflozine.
Ces différences ne sont pas considérées comme cliniquement significatives. Il n'existe aucune donnée clinique chez les patients atteints d'une insuffisance hépatique sévère (classe C de Child-Pugh).
Patients âgés
L'âge n'a pas eu d'effet cliniquement significatif sur la pharmacocinétique de la canagliflozine, d'après une analyse pharmacocinétique en population (voir rubriques Posologie et mode d'administration, Mises en garde spéciales et précautions d'emploi et Effets indésirables).
Population pédiatrique
Une étude pédiatrique de phase 1 a analysé la pharmacocinétique et la pharmacodynamique de la canagliflozine chez l'enfant et l'adolescent âgés de 10 à 18 ans atteints de diabète de type 2. Les réponses pharmacocinétiques et pharmacodynamiques observées étaient cohérentes avec celles trouvées chez le sujet adulte.
Autres populations particulières
Pharmacogénétique
UGT1A9 et UGT2B4 sont toutes deux soumises au polymorphisme génétique.
Dans une analyse poolée de données cliniques, des augmentations de
l'ASC (aire sous la courbe) de la canagliflozine de 26 % ont été
observées chez des sujets porteurs de l'allèle UGT1A9*1/*3 et 18 % chez
les sujets porteurs de l'allèle UGT2B4*2/*2. Ces augmentations de
l'exposition à la canagliflozine ne sont pas censées être cliniquement
pertinentes. L'influence de l'homozygotie (UGT1A9*3/*3, fréquence <
0,1%) est probablement plus marquée, mais n'a pas été étudiée.
Sexe, origine ethnique ou indice de masse corporelle n'ont aucun effet cliniquement significatif sur la pharmacocinétique de la canagliflozine d'après une analyse pharmacocinétique en population.
La canagliflozine n'a aucun effet ou qu'un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Cependant, les patients doivent être informés du risque d'hypoglycémie lorsque la canagliflozine est utilisée en association à l'insuline ou un sécrétagogue de l'insuline, ainsi que du risque accru d'effets indésirables liés à la déplétion volémique, comme les sensations vertigineuses posturales (voir rubriques Posologie et mode d'administration, Mises en garde spéciales et précautions d'emploi et Effets indésirables).
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée et génotoxicité n'ont pas révélé de risque particulier pour l'homme.
La canagliflozine n'a montré aucun effet sur la fertilité et le développement embryonnaire précoce chez le rat, à des expositions jusqu'à 19 fois l'exposition humaine à la dose maximale recommandée chez l'Homme.
Dans une étude du développement embryo-fœtal chez le rat, des retards d'ossification des métatarses ont été observés à des expositions systémiques 73 fois et 19 fois plus élevées que les expositions cliniques aux doses de 100 mg et 300 mg. On ne sait pas si les retards d'ossification peuvent être attribués aux effets de la canagliflozine sur l'homéostase calcique observés chez les rats adultes. Des retards d'ossification ont également été observés pour l'association canagliflozine et metformine, qui étaient plus importants que pour la metformine seule à des expositions de canagliflozine 43 fois et 12 fois plus élevées que les expositions cliniques aux doses 100 mg et 300 mg.
Dans une étude de développement pré- et post-natal, la canagliflozine administrée à des rates depuis le 6ème jour de gestation jusqu'au 20ème jour d'allaitement, a entrainé une diminution du poids de la progéniture mâle et femelle, à des doses maternelles toxiques > 30 mg/kg/jour (exposition > 5,9 fois l'exposition humaine à la dose maximale recommandée chez l'Homme). La toxicité maternelle était limitée à une diminution de la prise de poids.
Lors d'une étude chez le rat juvénile, l'administration de canagliflozine du 21ème au 90ème jour postnatal n'a pas montré une sensibilité accrue par rapport aux effets observés chez les rats adultes. Cependant, une dilatation du pyélon a été remarquée avec la dose sans effet observable (NOEL = No Observed Effect Level), à des doses d'exposition 2,4 fois et 0,5 fois l'exposition clinique aux doses de 100 mg et 300 mg respectivement et ne s'est pas pleinement inversée pendant la période de récupération d'environ un mois. Les résultats rénaux persistants chez les rats juvéniles peuvent sans doute être attribués à la capacité réduite du rein en développement du rat à gérer l'augmentation des volumes d'urine liée à la canagliflozine, étant donné que la maturation fonctionnelle du rein du rat se poursuit jusqu'à l'âge de 6 semaines.
La canagliflozine n'a pas augmenté l'incidence des tumeurs chez les souris mâles et femelles lors d'une étude de 2 ans à des doses de 10, 30 et 100 mg/kg. La dose la plus élevée de 100 mg/kg correspondait à 14 fois la dose clinique de 300 mg d'après l'ASC. La canagliflozine a augmenté l'incidence de tumeurs testiculaires à cellules de Leydig chez les rats mâles pour toutes les doses testées (10, 30 et 100 mg/kg); la plus faible dose de 10 mg/kg correspond à environ 1,5 fois la dose clinique de 300 mg d'après l'ASC. Les doses supérieures de canagliflozine (100 mg/kg) chez les rats mâles et femelles ont augmenté l'incidence des phéochromocytomes et des tumeurs tubulaires rénales. D'après l'ASC, la NOEL de 30 mg/kg/jour pour les phéochromocytomes et les tumeurs tubulaires rénales correspond à environ 4,5 fois l'exposition à la dose clinique journalière de 300 mg. D'après les études mécanistiques cliniques et précliniques, les tumeurs à cellules de Leydig, les tumeurs tubulaires rénales et les phéochromocytomes sont considérés comme spécifiques au rat. Les tumeurs tubulaires rénales induites par la canagliflozine et les phéochromocytomes chez le rat semblent être dus à une malabsorption des glucides consécutive à l'action inhibitrice de la canagliflozine sur le SGLT1 dans les intestins de rats ; les études cliniques mécanistiques n'ont pas démontré de malabsorption des glucides chez l'homme à des doses de canagliflozine correspondant à 2 fois la dose clinique maximale recommandée. Les tumeurs à cellules de Leydig sont associées à une augmentation de l'hormone lutéinisante (LH), qui est un mécanisme connu de formation des tumeurs à cellules de Leydig chez le rat. Dans une étude clinique de 12 semaines, le taux de LH non stimulée n'a pas augmenté chez les patients masculins traités par canagliflozine.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Remboursement en fonction de l'indication (JO du 20/09/2023) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
- Traitement des patients adultes atteints de diabète de type 2
insuffisamment contrôlé par une monothérapie par la metformine ou un
sulfamide hypoglycémiant, en complément du régime alimentaire et de
l'exercice physique, et uniquement en association :
- en bithérapie uniquement avec la metformine ou avec un sulfamide hypoglycémiant ;
- en trithérapie uniquement avec la metformine et un sulfamide
hypoglycémiant ou avec la metformine et l'insuline ;
- Traitement des patients adultes atteints de diabète de type 2 avec
une maladie rénale chronique de stade 2 et 3 et une albuminurie en
association au traitement standard, comprenant un inhibiteur de
l'enzyme de conversion (IEC) ou un antagoniste du récepteur de
l'angiotensine 2 (ARA II).
En outre, la prise en charge de la spécialité, dans ces indications,
est subordonnée à une information adaptée et complète des patients sur
les symptômes liés à chacun de ces événements indésirables de type
amputation, acidocétose avec autosurveillance, infections génitales
dont gangrène de Fournier. Le patient doit être en mesure de réaliser
une cétonémie en autosurveillance en cas de survenue des signes
d'alerte d'acidocétose.
Comprimé pelliculé (comprimé).
Comprimé jaune, en forme de gélule, d'environ 11 mm de longueur, à libération immédiate et pelliculé, avec l'inscription « CFZ » sur une face et « 100 » sur l'autre face.
Plaquette thermoformée unitaire perforée en Polychlorure de vinyle/Aluminium (PVC/alu).
Présentation de 30 x 1 comprimés pelliculés
Chaque comprimé contient de l'hémihydrate de canagliflozine, équivalent à 100 mg de canagliflozine.
Excipient(s) à effet notoire:
Chaque comprimé contient 39,2 mg de lactose.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Lactose
Cellulose microcristalline
Hydroxypropylcellulose
Croscarmellose sodique
Stéarate de magnésium
Pelliculage
Alcool polyvinylique
Dioxyde de titane (E171)
Macrogol 3350
Talc
Oxyde de fer jaune (E172)